XX Gonadal Dysgenesis and Premature Ovarian Failure in 46,XX Individuals
Authors
INTRODUCTION
Phenotypic females with normal chromosomal complements (46,XX or 46,XY) may have gonadal failure. External appearance may be identical in the two situations, and gonadal findings similarly indistinguishable from 45,X gonadal dysgenesis (Turner syndrome). The general term XX gonadal dysgenesis can be applied to the 46,XX cases. Mosaicism with a coexisting 45,X line is always a possibility, but usually this seems to be excluded. In this chapter the genetic (Mendelian) course of ovarian failure in 46,XX women is discussed. A wide spectrum of disorders exists (Table 1). In this chapter premature ovarian failure, sometimes caused by the same factors that cause complete ovarian failure (primary amenorrhea), is also considered. The chapter "Ovarian Dysgenesis and Premature Ovarian Failure Caused by X Chromosomal Abnormalities" considers ovarian failure caused by abnormalities of the X chromosome (Turner syndrome), and begins with a review of reproductive embryology to which the reader may wish to refer. In the chapter ''Gonadal Dysgenesis in 46,XX Females (XY Gender-Reversal)", 46,XY females, who have complete failure of testicular development and sometimes present with similar appearance, are discussed.
| Table 1. The spectrum of XX gonadal dysgenesis* |
| Gonadotropin genes |
| Follicle-stimulating hormone (FSH) |
| Follicle-stimulating hormone receptor (FSHR) |
| Luteinizing-stimulating hormone (LH) |
| Luteinizing-stimulating hormone receptor (LHR) |
| XX gonadal dysgenesis without somatic anomalies |
| XX gonadal dysgenesis with anomalies |
| XX gonadal dysgenesis with neurosensory deafness (Perrault syndrome) |
| XX gonadal dysgenesis with cerebellar ataxia (heterogeneous) |
| XX gonadal dysgenesis in malformation syndromes (Table 3) |
| XX gonadal dysgenesis as one component of pleiotropic Mendelian disorders (Table 4) |
| Germ cell absence in both sexes (46,XX) |
| Without somatic anomalies1, 2 |
| Associated hypertension and deafness3 |
| Associated alopecia4 |
| Associated microcephaly and short stature5 |
| Adrenal and ovarian biosynthetic defects |
| 17α-hydroxylase (CYP17) |
| Aromatase (CYP19) |
| Disorders of carbohydrate |
| Galactosemia (galactose uridyl-transferase deficiency [GALT]) |
| Carbohydrate-deficient glycoprotein (phosphomannomutase deficiency [PMM2]) |
| Agonadia (46,XX cases) |
| Dynamic mutations (triplet repeat) |
| Fragile X (FRAXA) |
| Ovarian-specific autoimmune |
| Polyglandular autoimmune syndrome |
| Autosomal trisomies |
| Trisomy 13 |
| Trisomy 18 |
*In some conditions, premature ovarian failure (POF) is discussed as well. In other consitions, discussed later in this chapter, POF exists but gonandal dysgenesis is rare or unreported.
XX GONADAL DYSGENESIS
XX gonadal dysgenesis without anomalies
The form of 46,XX gonadal dysgenesis not associated with somatic anomalies is most often inherited in autosomal recessive fashion. Affected individuals are normal in stature (mean height, 165 cm);6 somatic features of Turner’s stigmata are usually absent. Presence of consanguinity pointed to autosomal recessive inheritance decades ago. Segregation analysis by the author and colleagues revealed the segregation ratio to be 0.16 for female sibs.7 Thus, two thirds of gonadal dysgenesis in 46,XX individuals is genetic. The nongenetic phenocopies could be caused by infection, infarction, infiltrative or autoimmune phenomena (see Chapter "Environmental Factors Causing Ovarian Failure in Humans" for further discussion).
Of clinical relevance is the variable expressivity in XX gonadal dysgenesis. In some families one sib has streak gonads, whereas another has primary amenorrhea and extreme ovarian hypoplasia (presence of a few oocytes).6, 7, 8, 9, 10, 11 If the mutant gene responsible for XX gonadal dysgenesis were capable of variable expression, that gene may be responsible for some sporadic cases of premature ovarian failure (POF).
The mechanism underlying failure of germ-cell persistence in XX gonadal dysgenesis without somatic anomalies is unknown, but there are many candidate genes. Any abnormality of meiosis could be manifested as ovarian failure and infertility in otherwise normal women. Other mechanisms by which mutant genes could act include interference with germ-cell migration, connective tissue abnormalities, or gonadotropin receptor abnormalities.
Identifying the specific autosomal genes responsible for the various forms of XX gonadal dysgenesis has been difficult. In many monogenic disorders efforts toward positional cloning are facilitated by the fortuitous family in which an autosomal translocation cosegregates with the disorder. Sporadic cases of XX gonadal dysgenesis have long been associated with reciprocal autosomal translocations, but there seems to be little reproducibility among autosomes involved. Genome-wide sib-pair analysis using polymorphic DNA markers (dinucleotide repeats; single nucleotide polymorphism) should theoretically identify chromosomal region(s) worthy of sequencing. Indeed, this method was used successfully in Finland to elucidate the form of XX gonadal dysgenesis caused by follicle-stimulating hormone receptor (FSHR) mutation, which is discussed below.10, 12 Construction of cDNA libraries of ovarian specific genes represent a more omnibus approach, combined with use of gene knockout technology in the mouse and sequencing of candidate genes in the human. Many mouse genes are of potential relevance (Table 2), and perturbations in their human homologues can be sought. Many of these genes may only manifest as ovarian failure, in contrast to predictions that other organ systems would also be disturbed. An example is bone morphogenetic protein (bmp). The conclusion is that ovarian meiosis is easily perturbed, secondarily leading to germ-cell failure.
Table 2. Selected mouse models of ovarian failure
Mutant Mouse/Transgene | Human Locus | Function | Mouse Phenotype |
Zinc finger X (zfx) knockout | Xp22.1-p21.3 | Transcription factor | Reduced number of oocytes, infertility, short stature [Luoh and colleagues13] |
Germ-cell deficient (gcd) unknown | Unknown | Unknown gene, generated by transgene insertion | Lack of germ cells in as early as day 11.5 of embryonic development [Pellas and colleagues14] |
White spotting (W) | 4p11-q12 | Tyrosine kinase receptor | Reduced pigmentation, anemia, lack of germ cells [Manové and colleagues15] |
Steel (Sl) | 12q22 | Mast cell growth factor | Reduced pigmentation, anemia, lack of germ cells [Matsui and colleagues16] |
Steroidogenic factor 1 (SF-1) knockout | 9q33 | Nuclear receptor factor | Ovarian agenesis, XY sex reversal, adrenal agenesis [Luo and colleagues17] |
mutS (E. coli) homolog 5 (MSH 5) | 6p21.3 | DNA mismatch repair | Absence of ovarian structure, normal oviducts, and uteri [Edelmann and colleagues18] |
Beta cell leukemia/lymphoma 2(Bcl-2) knockout | 18q21.3 | Cell death repressor protein | Accelerated atresia of primordial follicles [Ratts and colleagues19] |
Factor in germline α (Figα) | Unknown | Transcription factor | Females lack primordial follicles, males are normal [Soyal and colleagues20] |
Postnatal ovarian failure defects |
|
|
|
Growth differentiation factor 9 (GDF9) | 5 | Oocyte secreted growth factor | Block in prenatal follicle development, infertility [Dong and colleagues21] |
Follicle-stimulating hormone β | 11p13 | Glycoprotein hormone | Female infertility, block of folliculogenesis before antral stage subunit ββ knockout (FSHβ) [Kumar and colleagues22] |
FSH receptor (FSHR) knockout | 2p21-p16 | Hormone receptor | Female infertility, block in folliculogenesis before antral stage [Dierich and colleagues23] |
Estrogen receptor α (Erα) knockout | 11q12 | Hormone receptor | Absent corpora lutea, arrest of preovulatory follicle maturation [Lubahn and colleagues24] |
Connexin 37 knockout | 1p35.1 | Gap junction | Lack of Graafian follicles, failure to ovulate [Simon and colleagues25] |
mutL (E. coli) homologue 1 (MLH1) | 3p21.3 | DNA repair enzyme | Failure to complete meiosis II, normal estrous cycle [Edelmann and colleagues26] |
Zona matrix protein 3 (mZP3) knockout | 7q11.23 | Zona pellucida | Infertility, oocytes lack zona pellucida [Rankin and colleagues27] |
Nerve growth factor-induced gene NGFI- A knockout | 2q32.3-q33 | Transcription factor | Lack of corpora lutea, suppressed luteinizing hormone levels [Topilko and colleagues28] |
(From Simpson JL, Rajkovic A: Ovarian differentiation and gonadal failure. Am J Med Genet 89: 186, 1999.)
Perrault syndrome (XX gonadal dysgenesis with neurosensory deafness)
XX gonadal dysgenesis associated with neurosensory deafness is called Perrault syndrome.29 Perrault syndrome is inherited in autosomal recessive fashion.6, 30, 31, 32, 33 Endocrine features seem identical to XX gonadal dysgenesis without deafness. Attractive candidate genes exist in the connexin family, given that connexin 37 knock-outs cause germ cell failure in mice (Table 1) and given that connexin mutations are the most common explanation for deafness in humans.
Follicle stimulating hormone (FSH)
Mutations in follicle-stimulating hormone (FSH) are rare, but the two affected women who have been reported have predictably shown neither thelarche nor menarche. Affected women predictably have hypogonadotropic hypogonadisms. Matthews and colleagues34 described a homozygous 2 bp deletion (GT) in exon 3, condon 61. Layman et al.35 reported a woman in whom one allele showed a missense mutation (exon 3, condon 51), and the other allele showed a deletion (exon 3, condon 61).
Follicle-stimulating hormone receptor (FSHR) mutation
This disorder is found predominantly in Finland, where Aittomaki and colleagues10, 12 searched hospitals and cytogenetic laboratories throughout the country to identify 75 subjects having the XX gonadal dysgenesis phenotype. Diagnostic criteria consists of 46,XX women with primary or secondary amenorrhea whose serum FSH was 40 MIU/mL or more. The 75 included 57 sporadic cases and 18 cases having an affected relative (seven different families). Most resided in north central Finland, a more sparsely populated part of the country. Prevalence was 1 per 8300 liveborn Finnish females. This relatively high incidence is attributed to a founder effect. The segregation ratio of 0.23 for female sibs was almost identical with theoretical expectations for autosomal recessive inheritance. This is consistent with a high consanguinity rate (12%).
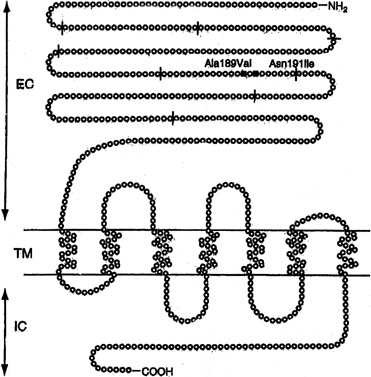
Sib pair analysis using polymorphic DNA markers first localized the gene to chromosome 2p, a region previously known to contain the genes for both the FSHR and the luteinizing hormone receptor (LHR). One specific mutation (Val to Ala) in exon 7 of the FSHR gene was found in six families10, 12 (Fig. 1). This cytosine-thymidine transition (called C566T) was later found in an additional six families.12, 36
|
Not all Finnish XX gonadal dysgenesis cases show the C566T mutation. Thus, the C566T-negative cases in Finland could represent the previously discussed condition XX gonadal dysgenesis with no somatic anomalies. Indeed, the C566T mutation is only rarely being detected in women with 46,XX ovarian failure who reside outside Finland. In the United States, Layman and colleagues37 failed to find a mutation in FSHR gene in 35 46,XX women having hypergonadotopic hypogonadism (15 with primary amenorrhea; 20 with secondary amenorrhea). Negative findings were reported in 46,XX POF or primary amenorrhea cases from Germany,38 Brazil,39 and Mexico.40 The last report analyzed all exons of FSHR. Liu and colleagues41 found no FSHR abnormalities in one multigenerational US POF family, four sporadic POF cases, and two other hypergonadotopic hypogonadism cases.
Aittomaki and colleagues36 compared the phenotype of Finnish C566T XX gonadal dysgenesis (C566T) with non-C566T XX gonadal dysgenesis. The former were more likely to have ovarian follicles by ultrasound. C566T XX gonadal dysgenesis thus showed at least some of the features gynecologists have long predicted for a gonadotropin resistance disorder (so-called Savage syndrome). In general, however, the phenotype found by Aittomaki10 was unexpected, bilateral streak gonads being found. FSHR (knockout) mice similarly show failure of oogenesis; thus, the necessity of FSH for progression of oogenesis is clear.42 Interestingly, FSH is less pivotal for spermatogenesis.43
Luteinizing hormone (LH)
In a single family in Brazil a homozygous luteinizing hormone (LH)β mutation was found in two hypogonadal males and their female sib. The mutation involved a 5’ splice site (IVs2 + IG→).35 The female sib underwent pubertal development but shortly thereafter experienced secondary amenorrhea.44
Luteinizing hormone receptor (46,XX)
Perturbation of the LH receptor gene (LHR) causes XX gonadal dysgenesis. The gene is 75 kd in length and consists of 17 exons. Located on 2p near FSHR, the first ten exons in LHR are extracellular; the last six are intracellular, and the 11th is transmembrane. LHR mutations have been reported predominately in 46,XY individuals, where the phenotype may extend to complete LH resistance and XY gender reversal (female). 46,XX cases are more rare than 46,XY cases, and have occurred only in sibships in which an affected 46,XY male had Leydig cell hypoplasia (XY gender reversal).
46,XX women with LHR mutations show oligomenorrhea or, less often, primary amenorrhea. Ovulation does not occur. Gametogenesis proceeds until the preovulatory stage, but not beyond. This is consistent with findings in the mouse knockout model.
Mutations in LHR are molecularly heterogeneous. Most mutations have been found in the transmembrane domain (exon). Latronico and colleagues45 reported primary amenorrhea in a 22-year-old woman whose family also included three affected males (46,XY), like the 46,XX sib homozygous for a nonsense mutation at codon 554 (Arg554ter). The resulting stop codon produced a truncated protein. The affected 46,XX female showed breast development but only a single episode of menstrual bleeding at age 20 years; LH was 37 mIU/mL, FSH 9 mIU/mL. The mutation reduced signal transduction activity of the LHR gene.
Toledo and colleagues46 studied the 46,XX female sib of two 46,XY affected males reported by Cramer and colleagues.47 The sister showed elevated gonadotropins but anatomically normal ovaries. The mutation was Ala593Pro. Two sisters reported by Laue and colleagues48 had the nonsense mutation cys545ter in exon 11; LHR function was lost. The father but not the mother had the mutation. The authors found a dominant negative effect, but more likely the mutant allele transmitted from the probably heterozygous mother has simply not yet been detected.
Cerebellar ataxia and XX gonadal dysgenesis
XX gonadal dysgenesis can be found in association with a heterogeneous group of cerebellar ataxias. The hereditary ataxias are confusing nosologically, principally because of ill-defined diagnostic criteria and lack of direct access to the cerebellum. Forms of ataxia characterized by hypogonadotropic hypogonadism also exist, but these are not considered here.
The association between hypergonadotropic hypogonadism and ataxia was first reported by Skre and colleagues.49 In one family, a 16-year old girl was affected, whereas in the other family there were three affected sisters. In the sporadic case and in one of the three sibs, ataxia was observed shortly after birth; in the two other sibs, age of onset occurred later in childhood. Cataracts were present in all individuals described by Skre and colleagues.49
Hypergonadotropic hypogonadism and ataxia was subsequently reported by De Michele and colleagues,50 Linssen and colleagues,51 Gottschalk and colleagues,52 Fryns and colleagues,53 Nishi and colleagues,33 and Amor and colleagues.54 In these reports the clinical features of ataxia have varied. Findings similar to those of Skre and colleagues49 were reported by De Michele and colleagues,50 Nishi and colleagues,33 and Amor and colleagues54; ataxia was usually not progressive. Mitochondrial enzymopathy was evident in one case reported by De Michele and colleagues,50 but in no other cases were mitochondrial studies conducted. Only Skre and colleagues49 observed cataracts and only Linssen and colleagues51 observed amelogenesis. Neurosensory deafness was reported by Amor and colleagues.54 Mental retardation is likewise variable.54
In conclusion, a single mutant gene is unlikely to explain every single case of XX gonadal dysgenesis and cerebella ataxia. However, not every family need be unique.
XX gonadal dysgenesis and multiple malformation syndromes
Several other pleiotropic genes cause XX gonadal dysgenesis and various somatic features. All are rare and perhaps even unique to a specific family. Table 3 lists these syndromes: XX gonadal dysgenesis, microcephaly, and arachnodactyly;55 XX gonadal dysgenesis, cardiomyopathy, blepharoptosis, and broad nasal bridge;54, 56 XX gonadal dysgenesis and epibulbar dermoid;57 and XX gonadal dysgenesis, short stature and metabolic acidosis.58 Assuming Mendelian etiology, these disorders are presumably autosomal recessive. However, subtle chromosomal rearrangements cannot be excluded.
Table 3. Malformation syndromes with 46,XX gonadal dysgenesis
Somatic Features | Reference | Etiology |
Cerebellar ataxia, sensori-neural deafness, other somatic features | Autosomal recessive, heterogeneous | |
Microcephaly, arachnodactyly | Maximilian and colleagues55 | Autosomal recessive |
Epibulbar dermoids | Quayle and Copeland57 | Autosomal recessive |
Short stature and metabolic acidosis | Autosomal recessive | |
Blepharophimosis-ptosis-epicanthus | Autosomal dominant (FOXL2) | |
Renal parenchymal disease, ovarian failure (46,XX Frasier syndrome) Wilms' tumor and genital ambiguity (Denys-Drash syndrome) different perturbations involving same gene (WT1) | Bailey and colleagues62 (Frasier syndrome) | Autosomal dominant (WT1) |
Dilated cardiomyopathy, mental retardation, bleparoptosis (Malouf syndrome) | Autosomal recessive | |
| Limb-mammary syndrome (LMS) ectrodactyly, ectodermal displasia, cleft lip/palate | Guazzarotti and colleagues64 | Autosomal dominant |
In all these syndromes, an underlying biologic question is whether the ostensibly pleiotropic gene(s) causes both somatic anomalies and ovarian failure? Alternatively, could the somatic and gonadal phenotypes merely reflect closely linked genes, that is, a contiguous gene syndrome? Irrespective of the afore-mentioned, do these purported genes play roles in normal ovarian differentiation, or does their perturbation merely cause ovarian failure secondary to generalized somatic disturbance?
Pleiotropic Mendelian disorders showing ovarian failure
Primary ovarian failure is observed frequently in well-established and not necessarily uncommon in Mendelian disorders. All are characterized by distinct somatic features (Table 4). Pleiotrophy for these mutant genes allows clear distinction from disorders previously discussed.
Table 4. Autosomal recessive disorders the phenotype of which is predominately somatic but in which ovarian failure is also observed
Disorder | Somatic Features | Ovarian Anomalies | Etiology |
Ataxia-telangiectasia | Cerebella ataxia, multiple telangiectasias (eyes, ears, flexa surface of extremities), immunodeficiency, chromosomal breakage, malignancy, x-ray hypersensitivity | “Complete absence of ovaries”, “absence of primary follicles” [Zadik and colleagues;65 Waldmann and colleagues66] | |
Carbohydrate-deficient glycoprotein syndrome, type 1 (phosphomannonutase deficiency) [Matthijs and colleagues67] | Neurologic abnormalities (e.g., unscheduled eye movements), ataxia, hypotonial/hyporeflexia strokes, joint cartractures | Ovarian failure (hypogonadism) [Kristiansson and colleagues68] | Autosomal recessive |
Cockayne Syndrome [Nance and Berry69] | Dwarfism, microcephaly, mental retardation, pigmentary retinopathy and photosensitivity, premature senility. Sensitivity to ultraviolet light | Ovarian atrophy and fibrosis [Sugarman and colleagues70] | Autosomal recessive |
Galactosemia (galactose-l-phosphate uridyl transferase deficiency) (GALT) | Hepatic failure with cirrhosis, renal failure, cataracts, cardiac failure | Ovarian failure with streak gonads [Kaufman and colleagues;71 Waggoner and colleagues;72 Levy and colleagues73] | Autosomal recessive |
Martsolf syndrome [Martsolf and colleagues74] | Short stature, microbrachycephaly, cataracts, abnormal facies with relative prognathism due to maxillary hypoplasia | “primary hypogonadism” [Harbord and colleagues;75 Hennekam and colleagues76] | Autosomal recessive |
Nijmegen syndrome [Weemaes and colleagues77] | Chromosomal instability, immunodeficiency, hypersensitiviy to ionizing radiation, malignancy | Ovarian failure (primary) [Conley and colleagues;78 Chrzanowska and colleagues79] | Autosomal recessive |
Rothmund-Thompson syndrome [Hall and colleagues80] | Skin abnormalities (telangiectasia, erythrema irregular pigmentation), short statue, cataracts, sparse hair, small hands and feet, mental retardation, osteosarcoma | Ovarian failure (primary hypogonadism or delayed puberty) [Starr and colleagues81] | Autosomal recessive |
Werner syndrome [Goto and colleagues82] | Short stature, premature senility, skin changes (scleroderma) | Ovarian failure [Goto and colleagues82] | Autosomal recessive |
Of special note is Denys-Drash syndrome and Frasier syndrome, which are caused by mutations in WT-1 (Wilms’ tumor-1), a gene located on 11p. WT-1 mutations can cause either 46,XY gender reversal or 46,XY genital ambiguity. One 46,XX individual with Frasier syndrome has been reported.62 This woman manifested not only the renal parenchymal disease characteristic of Frasier syndrome, but also primary amenorrhea and ovarian failure. Gonadal failure in 46,XX Frasier syndrome could easily pass unappreciated if the primary amenorrhea is assumed to occur secondary to azotemia.
Germ-cell failure in male (46,XY) and female (46,XX) sibs
In several sibships, male and female sibs have shown germinal cell failure. Affected 46,XX females showed streak gonads, whereas 46,XY affected males showed germ-cell aplasia (Sertoli-cell-only syndrome). In two families, parents were consanguineous, and in each there were no somatic anomalies.1, 2 In three other families (see Table 1), characteristic somatic anomalies suggested distinct entities. Hamet and colleagues3 observed germ-cell failure, hypertension, and deafness; Al-Awadi and colleagues4 found germ-cell failure and alopecia; Mikati and colleagues5 reported germ-cell failure, microcephaly, and short stature.
In each of these families a single autosomal gene is presumed to affect germ cell development deleteriously in both sexes. This gene(s) could act either at a specific site common to early germ cell development or exert its effect through meiotic breakdown. Elucidating such genes could help explain normal germ cell development. Indeed, a variety of attractive candidate genes exist in mouse and Drosphila (see Table 2). An example is gcd, a mouse mutant in which germ cells are deficient in both males and females. See the chapter, "Cytogenetic, Teratogenic, and Miscellaneous Other Disorders Causing Male Pseudohermaphroditism or Germ-Cell Failure in Both Males (46,XY) and Females (46,XX)".
Inborn errors of metabolism
Galactosemia is caused by deficiency of galactose 1-phosphate uridyl transferase (GALT). The gene is located on 9p. Long-recognized features included renal, hepatic, and ocular defects. Kaufman and colleagues71 reported POF in 12 of 18 galactosemic women, and Waggoner and colleagues72 observed ovarian failure in eight of 47 females. Pathogenesis was presumed to involve galactose toxicity during infancy or childhood; elevated fetal levels of toxic metabolites should be cleared rapidly in utero by maternal enzymes. Consistent with this hypothesis, is that a neonate with galactosemia showed normal ovarian histology.73
Given the clinical severity of galactosemia and the absolute necessity for dietary treatment during childhood to prevent mental retardation, it seems highly unlikely that previously undiagnosed galactosemia would prove to be the cause of ovarian failure in women presenting solely with primary amenorrhea or POF. Of greater relevance to gynecology, therefore, was the report in 1989 by Cramer and colleagues83 that GALT heterozygotes were at increased risk for POF.83 However, the same author later failed to observe GALT abnormalities in another sample of women with early menopause,47 and Kaufman and colleagues84 likewise failed to confirm the observation. Moreover, not all homozygotes for human galactosemia are abnormal, nor are transgenic mice in which GALT is inactivated.85
Carbohydrate-deficient glycoprotein intrauterine contraceptives (phosphomannomutase deficiency [PMM2])
In type 1 carbohydrate-deficient-glycoprotein (CDG) deficiency, mannose-6-phosphate cannot be converted to mannose-1-phosphate. Thus, the lipid-linked mannose-containing oligosaccharides needed for secretory glycoproteins are lacking. The CDG gene is located on 16p13. The molecular pathogenesis is usually a missense mutation.86
The wide spectrum of neurologic abnormalities includes hypotonia, hyperreflexia, unprovoked eye movements, ataxia, joint contractions, epilepsy, and stroke-like episodes.67 Subcutaneous fat deposits, hepatomegaly, cardiomyopathy, pericardial effusion, and factor XI (clotting) deficiency develop.
Of interest here is that ovarian failure occurs; the ovaries are devoid of follicular activity.68, 87
Deficiency of 17α-hydroxylase/17,20-desmolase (lyase) deficiency (CYP17)
Sex steroid synthesis requires intact adrenal and gonadal biosynthetic pathways. Various gene products (enzymes) are necessary to convert cholesterol to testosterone and androstenedione and, hence, estrogens. The various enzyme blocks have varying but predictable consequences, depending on their site in the biosynthetic pathway (Fig. 2). The most common adrenal biosynthetic problem is involving deficiency of 21- or 11β-hydroxylase, either of which causes pseudohermaphroditism. These disorders cause genital ambiguity because of virilization, but need not be considered in the differential diagnosis of XX gonadal dysgenesis.
|
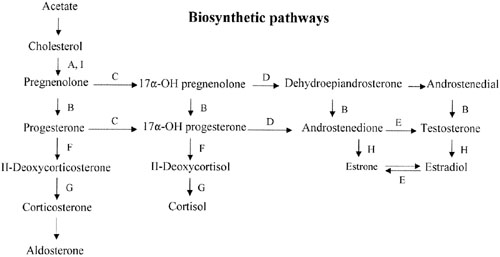
If the cytochrome P450 enzyme 17α-hydroxylase/17-20-lyase is deficient, pregnenolone cannot be converted to 17α-hydroxy-pregnenolone. If the enzyme defect were complete, cortisol, androstenedione, testosterone, and estrogens could not be synthesized; however, 11-deoxycorticosterone and corticosterone could. With compensatory increases in adrenocorticotropic hormone (ACTH secretion), 11-deoxycorticosterone and corticosterone result in hypernatremia, hypokalemia, and hypervolemia. Hypertension occurs. Aldosterone is decreased, presumably because hypervolemia suppresses the renin-angiotensin system.
Females (46,XX) with 17α-hydroxylase deficiency have normal external genitalia, but at puberty fail to undergo normal secondary sexual development (primary amenorrhea). Affected males (46,XY) usually have genital ambiguity (male pseudohermaphroditism) because partial expression of the gene produced some androgens. Affected females are ordinarily encountered in differential diagnosis of XX gonadal dysgenesis. Hypertension is the major distinguishing clue. Oocytes appear incapable of spontaneously exceeding a diameter greater than 2.5 mm,88 but ovaries nonetheless respond to exogenous gonadotropins.89
17α-hydroxylase deficiency is inherited in autosomal recessive fashion. The gene (CYP17) is located on 10q24-25, and the gene product is a cytochrome P450 enzyme. More than 20 different mutations have been identified in CYP17, scattered among the eight exons. Mutations include missense mutations, duplications, deletions, and premature protein truncation.90 Most mutations have been observed in only a single family, yet another example of molecular hetereogeneity. An exception exists in Mennonites of Dutch origin, where a four-base duplication in exon 8 accounts for most cases.91 This founder mutation originated in Friesland.
This single gene (and enzyme) is responsible for both 17α-hydroxylase and 17,20-desmolase (lyase) actions (see Fig. 2). A few patients having deficiency of both 17α-hydroxylase and 17,20 lyase activities have been analyzed, with mutations different from those only showing deficient 17α-hydroxylase activity being found.92, 93 Transfection experiments show that only 5% of 17α-hydroxylase activity is sufficient for the estrogen production necessary for normal secondary sexual characteristics in a 46,XX individual; however, 25% of enzyme activity is necessary to virilize external genitalia in males.90, 92 Targeted mutagenesis in the rat gene indicates that mutations closer to the 5′ end are more deleterious.
Aromatase mutations (CYP19)
Conversion of androgens (Δ4-androstenedione) to estrogens (estrone) requires cytochrome P-450 aromatase, an enzyme product of a 40-kb gene located on chromosome 15q21.1.94 Deficiency of the aromatase enzyme in 46,XX individuals is most often associated with clitoral hypertrophy or genital ambiguity, but 46,XX aromatase deficiency may present as primary amenorrhea in phenotypic females.
Ito and colleagues95 reported aromatase mutation (CYP19) in an 18-year-old 46,XX Japanese woman having primary amenorrhea and cystic ovaries. Compound heterozygosity, existed for two point mutations in exon 10. The mutant protein had no activity. Conte and colleagues96 reported aromatase deficiency in a 46,XX woman presenting with primary amenorrhea, elevated gonadatropins, and ovarian cysts. Compound heterozygosity also existed for two mutations in exon 10. One mutation was ′C1303T (cysteine rather than arginine); the other was ′G1310A (tyrosine rather than cysteine).
Mullis and colleagues97 reported clitoral enlargement at puberty. No breast development occurred. FSH was elevated; estrone and estradiol were decreased. Multiple ovarian follicular cysts were evident. Hormonal (estrogen and progesterone) therapy produced a growth spurt, breast development, menarche, and decreased numbers of follicular cysts. Compound heterozygosity existed in CYP19.
46,XX agonadia
Agonadia usually occurs in 46,XY individuals but, rare 46,XX cases exist. As in 46,XY agonadia, gonads are absent, in 46,XX agonadia. This contrasts with persistence in the form of streaks (gonadal dysgenesis). External genitalia are abnormal but female-like; no more than rudimentary Müllerian or Wolffian derivatives are present. External genitalia usually consist of a phallus about the size of a clitoris, underdeveloped labia majora, and nearly complete fusion of labioscrotal folds. Thus, external genitalia are nearly or somewhat female in appearance. In approximately one half of cases, somatic anomalies coexist: craniofacial anomalies, vertebral anomalies, and mental retardation.
Agonadia must be considered in the differential diagnosis of 46,XX primary amenorrhea. The 46,XX agonadia cases reported by Duck and colleagues98 and Levinson and colleagues99 were sporadic. Mendonca and colleagues100 reported agonadia without somatic anomalies in phenotypic sibs having unlike chromosomal complements (46,XY and 46,XX). Kennerknecht and colleagues101 reported agonadism, hypoplasia of the pulmonary artery and lung, and dextrocardia in XX and XY sibs.
PREMATURE OVARIAN FAILURE
Premature ovarian failure102, 103, 104, 105 can result from nongenetic causes as well as from several different genetic causes. Genetic or familial POF can be stratified into five general etiologies: (1) autosomal rearrangements; (2) X-chromosomal abnormalities; (3) autosomal recessive genes (sometimes the same genes resulting in the various forms of XX gonadal dysgenesis; see above); (4) autosomal chromosomal rearrangements; and (5) autosomal dominant genes including those action of which may or may not be restricted to POF. In addition, the possibility exists that some cases of POF merely represent the lower end of the normal population with respect to ovarian number and function (i.e., early but normally executed menopause). We consider this concept first.
There is broad overlap between complete ovarian failure (gonadal dysgenesis) and the much more common (1%) prevalance premature ovarian failure (POF). The usual definition is ovarian failure before age 40 years. Perturbations of a given gene can cause either complete or partial (premature) ovarian failure. Thus, there is considerable redundancy in discussing POF per se. The disorders covered here in detail are those that primarily present as POF, not as primary amenorrhea.
Premature ovarian failure as normal continuous variation: heritable early menopause
Oocyte number (reservoir) could be low in some women simply on statistical (stochastic) grounds. Normal distribution exists for all common anatomic traits (e.g., height), and this principle must apply to oocyte number and reservoir at birth. Different rodent strains show characteristic breeding duration, implying genetic control over either the rate of oocyte depletion or the number of oocytes initially present. That a normal distribution of germ cell number exists in ostensibly normal females is thus well established in animals, but difficult to prove in humans. Nonetheless, some ostensibly normal (menstruating) women should have a decreased oocyte reservoir or increased oocyte attrition on a genetic basis, analogous to animal models.
In humans, a genetic basis for the above can also be presumed by analogy to the heritability of age at human menopause, a characteristic that clearly shows familiar tendencies. However, determining heritability of age of menopause in humans is complicated because of iatrogenic factors (e.g., hysterectomy) and other confounders (e.g., presence of leiomyomata). Cramer and colleagues47 took these confounders into account in a case–control study of 10,606 U.S. women. Women with an early menopause (40–45 years) were age-matched with controls who were either still menstruating or who had experienced menopause after age 46. Of 129 early menopause cases, 37.5% had a similarly affected mother, sister, aunt, or grandmother. Only 9% of controls had such a relative (odds ratio after adjustment 6.1, 95% confidence interval [CI] 3.9–9.4). As predicted on the basis of polygenic inheritance, the odds ratio was greatest (9.1) for sisters and greater when menopause occurred prior to 40 years. Incidentally, frequency of galactose 1-phosphate uridyltransferase (GALT) variants (N314D or Q188R) did not deviate from that expected in early menopause cases, in contrast to earlier studies by the same authors.83 The results of Cramer and colleagues47 were confirmed by Torgerson and colleagues,106 who studied women undergoing menopause during the 5-year centile ages 45–49 years. The likelihood was increased that menopause would occur in a similar 5-year centile in their daughters.
Twin studies have confirmed heritability of age at menopause.107 Snieder and colleagues108 studied 275 monozygotic (MZ) and 353 dizygotic (DZ) UK twin pairs. For age at menopause, correlation (r) was 0.58 for MZ and 0.39 DZ twins; heritability (h7) was calculated to be 63%. The study of Treloar and colleagues109 involved 466 MZ and 262 DZ Australian twin pairs. For age at menopause the correlation (r) was 0.49–0.57 for MZ and 0.31–0.33 for DZ twin pairs. Differences between MZ and DZ held when iatrogenic causes of menopause was taken into account.
Autosomal chromosomal rearrangements
Autosomal chromosomal rearrangements can also lead to premature ovarian failure with many different reciprocal translocations involved. The underlying mechanism is presumably meiotic breakdown. About 2% of azoospermic or oligospermic men who present for intracytoplasmic sperm injection (ICSI) but are otherwise clinically normal show balanced autosomal translocations.110, 111
Approximately 2% of their ostensibly normal female partners also show balanced translocations. A relation thus also exists between balanced translocations and infertility in women, but lack of a readily assayed end point to pinpoint infertility makes studies more difficult in females than in men. The frequency in infertile women having normal male partners has not been crisply defined because of pronounced biases of ascertainment in almost all series. However, the prevalence is surely at least 2% and probably higher. The same translocation almost certainly predisposes to POF or early menopause.
Detecting individuals with a chromosomal rearrangement is important not least as an explanation for their offspring being at risk for unbalanced chromosomal abnormalities.
Autosomal dominant premature ovarian failure
Autosomal dominant familial tendencies per se have long been recognized in cytogenetically normal women with POF who have no other clinical abnormalities. Thus, discussion here excludes X-abnormalities [del(Xp) and (Xq)] and the syndrome blepharophimosis-ptosis-epicanthus as a result of FOXL2 perturbations. Other samples of women with POF report high frequencies of associated abnormalities, such as Kim and colleagues;112 22 of 119 women with POF (18.5%) were hypothyroid, and three had Addison’s disease. Although this seems atypical, for many years it was widely assumed that familial tendencies in POF merely reflected autoimmune phenomena. In women with POF, antibodies against adrenal,113, 114, 115 thyroid,116 and pancreas114 may be detected;117 however, autoimmune factors are not always or even often observed. Familial tendencies in the absence of autoimmune findings are more common. This was observed decades ago. Coulam and colleagues118 reported POF in sibs who had an affected mother and aunt. Affected individuals in more than one generation were reported by Starup and Sele,119 Austin and colleagues,120 and Mattison and colleagues.121 In none of these families were autoimmune phenomena observed.
In Italy, Testa and colleagues122 and Vegetti and colleagues123 systematically studied women first with POF (menopause at younger than 40 years of age) recruited from a large northern Italian population. After excluding ten cases with known etiologies (five chromosomal, three prior ovarian surgery, one prior chemotherapy, one galactosemia), 71 probands remained. Of the 71, 22 (31%) had other affected relatives (Table 5). An expanded sample of 130 cases found 28.5% familial cases.124 Patterns of inheritance observed were consistent with autosomal or X-linked dominant inheritance; transmission through both maternal and paternal lineage was observed. Among 30 other women experiencing early menopause (40–50 years) half showed other affected relatives. There is further evidence that POF (younger than 40 years) and early menopause (menopause at 40 to 50 years) represent the same phenomena. The two different phenotypes have been observed within a single kindred, transmission through either a paternal or maternal relative.
Table 5. Frequency of heritable premature ovarian failure in an Italian referral sample
Known etiology (n = 10) | |
Abnormal chromosomal | 5 |
Prior ovarian surgery | 3 |
Prior chemotherapy | 1 |
Galactocemia | 1 |
Idiopathic (n = 71) | |
No family history | 49 |
Family history | 22 (31%) |
| Total | 81 |
Data of Vegetti and colleagues.125
Pathogenic mechanisms for autosomal dominant premature ovarian failure include decreased number of primordial follicles or human homologous of mouse cited in Table 2. Nongenetic etiologies (phenocopies) might include infiltrative disease (e.g., sarcoidosis), toxins, or autoimmune phenomenon. Environmental causes are unlikely to explain intergenerational familial aggregates.
Autosomal genes of known endocrine function
Disturbances of these genes usually present as primary amenorrhea and thus have already been discussed.
Follicle stimulating hormone (FSH) – see above.
FSH receptor – see above.
Luteinizing hormone (LH) – see above.
LH receptor (LHR) – see above.
Inhibin α (INHα) – inhibins (INH) are heterodimeric glycoproteins, synthesized by granulose cells. Inhibin consists of an α subunit and one of the two B submits (BA and BB), producing INHA and INHB, respectively. These genes exert negative feedback inhibition on FSH. By contrast, activins enhance FSH secretions. In premature ovarian failure serum inhibin is low and FSH is elevated. Elevated FSH and low inhibin indicates reproductive aging. Preturbations of inhibins should cause ovarian failure.
Three studies have shown an association between POF and a particular INHα missense mutation – G769A. Shelling et al.126 found G769A in three of 43 New Zealand POF patients (7%); only one of 150 controls (0.7%), showed G769A. However, the fact that the mother of the three G769A cases had the same perturbation and was clinically normal casts doubt on causality. Marozzi et al.127 found G769A in seven of 157 Italian POF cases, three of 12 primary amenorrhea cases, and none of 36 early menopause (40–45 years) women. Dixit et al.105 reported that familial POF cases were relatively more likely to show G769A (9/80 cases) than were sporadic cases.
Concluding causation on the basis of gene association studies per se is never possible because of pitfalls like ethnic stratification, and there is reason to be skeptical. The G769A transition may not be pivotal, given that normal individuals also have the G769A transition and given that individuals with G769A may be abnormal even in a family in which a G769A relative has POF. However, INHα remains a attractive candidate gene, and in vitro functional studies have revealed positive findings.
Cytochrome P450 17 (CYP17) (17α-hydroxylase/17,20 desmolase deficiency) – see above.
Of specific relevance to POF is that ovaries are hypoplastic and may be streak-like in appearance. Oocytes appear incapable of reaching diameters of more than 2.5mm.128 However, ovarian stimulation can produce oocytes capable of fertilization in vitro.
Cytochrome P450 19 (CYP19) (aromatase) – see above.
Other genes required for oogenesis
NOBOX
NOBOX (Newborn Ovary homeoBOX gene) encodes a homeobox transcriptional regulator.104 NOBOX is ocyte-specific and expressed from the primordial follicle through metaphase II. Female null mice (knockout) show ovarian failure, whereas males are normal. One study failed to show NOBOX perturbations in 30 Japanese women,129 but our group later found two novel missense mutations (Arg355His and Arg360Gln) among 96 caucasian POF cases.130 Arg355His was found in 278 controls. A functional effect was shown by electrophoretic mobility shift array (EMSA); Arg355His disrupted binding of the NOBOX homeodomain to DNA, showing capacity for a dominant negative effect.
LHX8 (LIM DNA-BINDING PROTEINS)
LIM homeobox genes encode DNA-binding proteins, serving as transcriptional factors that play critical roles in embryonic differentiation. LHX8 transcripts localize to mouse oocytes from germ cells through antral follicles. Null mice lack germ cells.131
Our group sequenced LHX8 in 95 caucasian women with POF132 and found no abnormalities. Additional studies are necessary before it can be concluded that perturbation of this gene can cause POF in humans.
NANOS3 (RNA-BINDING PROTEINS)
NANOS3 is a RNA-binding protein. Both female and male knockout mice are infertile.133 Human NANOS3 gene consists of two exons and is expressed in germ cells.
Our group sought NANOS3 perturbations in 80 Chinese and 88 American caucasians, but the only sequence varient found was a synonymous nucleotide substitution that had already been reported.134
GDF9 (GROWTH DIFFERENTIATION FACTOR 9) AND TRANSFORMING GROWTH FACTORS
Transforming growth factor (TGF) superfamily genes are also transcription factors, and encoded by both sex chromosomes (Bone morphogenetic protein 15, BMP15) and autosomes. Located on chromosome 5 (5q31.1), GDF9 has three subunits: signal peptide, prodomain, and mature region. This arrangement is similar to other TGF-β family members. BMP15 and CDF9 gene products can form heterodimers.
Expressed in oocytes, GDF9 is believed to play an essential role in both early and late folliculogenesis.135 GDF9 protein promotes cumulus expansion; its suppression (double-stranded interfering RNA) prevents cumulus expansion. In null mice oogenesis does not proceed, mutations thus causing disorders of follicle function rather than primordial germ cell formation. Immunization against GDF9 in sheep disrupts early folliculogenesis and leads to the absence of normal follicles beyond the primordial stage.
Human POF cases have been investigated in several populations for GDF9 perturbations. Of 127 Indian women with POF, Dixit et al.134 found five to have the missense mutation A199C and two to have a G646A mutation. All mutations were heterozygous, occurring in the preprotein region. Our group more recently surveyed 61 American women with POF. A single woman showed a heterozygous perturbation (c.307C>T) that resulted in proline being replaced with serine (P103S).136 Supporting plausibility of a deleterious effect was that the perturbation involved a conserved region and involved replacement of a hydrophobic amino acid (proline) with a hydrophilic amino acid (serine) residue. Among 100 Chinese POF cases, we also found a novel propeptide mutation (Thr238Ala) not present in 96 Chinese controls.137 Again, substitution of the hydrophobic alanine for the hydrophilic threonine could be deleterious. The plausibility of a deleterious GDF9 heterozygous change rests on the assumption that a dominant negative mutation exists.
GPR3 (G PROTEIN RECEPTOR 3 and G PROTEINS)
G proteins (GPs) and their receptors (GPRs) constitute a family of regulatory proteins involved in intracellular and intercellular transduction. Disparate stimuli (e.g., hormones, light glycoproteins) all exert conformational changes on GPRs to allow binding to GPs. GPs and GPRs are crucial for reproductive function. The oocyte-specific G-stimulating protein-coupled receptor GPR3 is a known factor in maintaining meiotic arrest in the mouse oocyte. Female mice lacking GPR3 develop premature ovarian aging as a result of spontaneous resumption of meiosis in antral follicles, independent of the LH surge.138 Oocyte attrition results.
Our group sought GPR3 perturbations in caucasian women with POF; none of the 82 showed perturbations of significance.138 A single subject showed heteroduplex formation due to a heterozygous nucleotide substitution, C to A at position 51 (c.51C>A). However, this substitution does not alter the amino acid sequence, and had already been identified in normal women. Although GPR3 mutations do not appear to be a common explanation for POF in North American caucasians, studies in other populations are needed.
ANGIOTENSIN II TYPE 2 (AT2) RECEPTOR
This gene is expressed in fetal tissue and in a diverse group of disease states. Atretic granulosa cells express the gene in rodents. The gene is located on Xq22-23, a region of known significance, so-called POF2. Katsuya et al.139 studied two families, in each of which sibs each had POF; no AT2 mutations were found.
PIEBALDISM (KIT)
Piebaldism is an autosomal dominant disorder characterized by patches of depigmentation (white) skin and hair.140 In contrast to vitiligo, piebaldism is present at birth and progressive. Hyperpigmented macules are present on both normal and depigmented skin. Some cases are caused by mutations of KIT, an autosomal (4q12) gene that encodes for the tyrosine kinase transmembrane regulator for mast/stem cell growth factor.140 Mutations exerting causality are undisputable, and include missense, nonsense, deletions, insertions, and splice junction mutations.
The c-kit receptor and its ligand (KL) cause germ cell loss in mice if perturbed, corresponding to the white spotting (W) and steel (SI) loci. Human KIT is thus a good candidate gene for POF. Shibanuma et al.141 studied 40 women with unexplained POF, sequencing the entire coding region. One synonymous missense mutation was found, but this was not a plausible explanation for POF.
ANTIMULLERIAN HORMONE (AMH) AND RECEPTOR (AMHR)
In addition to its role in Müllerian duct regression in males, AMH is an oocyte inhibitor in the rat. The locus for AMH is on 19p, whereas that for the AMH receptor is 12q. The murine knockout model shows early depletion of primordial follicles.142 Wang et al.143 failed to find plausible perturbations in 16 POF cases in humans.
RET FINGER-LIKE PROTEIN 4 (RFLP4)
This RING finger-line protein is expressed exclusively in murine oocytes, functioning as an E3 ubiquitin protein ligase. Ubiquitin ligase regulates protein degradation. Human RFLP4 is located on 19q13.4, and has been shown to interact with oocyte proteins of the ubiquitin-protease degredation pathway.144 Suzumori et al.103 found no mutations 'in Japanese POF patients with 46,XX POF'.
Pleiotropic genes causing premature ovarian failure
BLEPHAROPHIMOSIS-PTOSIS-EPICANTHUS (FORKHEAD TRANSCRIPTION FACTORS) (FOXL2)
Type 1 blepharophimosis-ptosis-epicanthus syndrome (BPES) is an autosomal dominant disorder in which POF coexists with a number of eyelid abnormalities.145 Perturbations of forkhead transcription factor FOXL2 are the cause. In BPES type 1, FOXL2 mutations of unassailable significance (e.g., stop condon and truncated protein) have been found.146 In the absence of eyelid abnormalities, however, mutations in this single exon gene are uncommon explanations for POF. De Baere and colleagues147 found no FOXL2 mutations in 30 POF patients with normal eyelids; Harris and co-workers148 found two mutations among 70 POF cases; Watkins et al.149 found eight mutations among 233 cases (3.4%).
BPE is an autosomal dominant multiple malformation syndrome associated with ovarian failure. Usually POF exists, not complete ovarian failure.60, 150 In one unusual case, Fraser and colleagues151 reported that the ovaries of an affected individual were unresponsive to gonadatropins.
The gene is localized to chromosome 3 (3q22→24)152 a region that contains no obvious candidate gene.153 Positional cloning revealed mutation in the winged helix/forkhead transcription factor gene (FOXL2), yielding a truncated protein61 (Table 6). This indicates that the mechanism of gene action is haplo-insufficiency. Mutations observed included three stop codons (X) and a 17-base pair duplication causing a frameshift and, hence, truncated gene product. Postulated FOXL2 gene function includes inhibition of proapoptotic genes such as tumor necrosis factor (TNF)-α, thus, sustaining follicle viability.
Table 6. FOXL2 mutations in blepharophimosis-ptosis epicanthus syndrome (BPES, type 1)
Family | Nucleotide | Amino Acid |
1 | 892C→T | Q219X |
2 | 848G-A | W204X |
3 | 1092-1108dup | H291fsX361 |
4 | 737T→A; 738C→A | F167X |
Data of Crisponi and colleagues.61
The gene is expressed in mesenchyma of mouse eyelids and in adult ovarian follides, consistent with phenotype.
FOX03A
Other forkhead transcription genes cause ovarian follicular depletion when genes are knocked out (null) in mice. FOX03A regulates G1/S transition in granulosa cells. Watkins et al.149 sought mutations in the coding region of FOX03A. Of 60 POF cases, two showed potentially significant mutations not present in controls. One mutation was a single heterozygous mutation in a Slovenian woman. The non-conserved amino acid change (Ser421Leu) was potentially capable of inducing a conformational protein change. The other mutation found was Arg506His in a New Zealand woman.
FOX01A
Watkins et al.149 found a single conservative change (P84L) in FOX01A among 90 POF cases. The patient was of Slovenian descent.
FRAGILE X SYNDROME AND EXPANSION OF THE TRIPLET NUCLEOTIDE REPEATS (CGG)
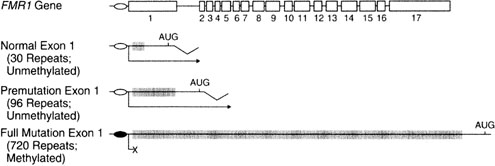
"Fragile" X syndrome, caused by mutation of the FMRI gene on Xq27 refers to a tendency toward chromosomal breakage when affected cells are cultured in folic acid-deficient media. Various fragile sites exist in humans, but only FRAXA and FRAXE in particular are relevant.
In FRAXA, males show mental retardation, characteristic facial features, and large testes. The molecular basis involves repetition of the triplet repeat CGG (CGG)n 230 times or more (Fig. 3). Ordinarily, the normal number of repeats in males is only 29–30. When heterozygous females show 55–200 repeats, a premutation is said to be present. During female (but not male) meiosis the number of triplet repeats may increase (expand). A woman with a FRAXA premutation may have an affected son if the number of CGG repeats on the X transmitted to her son were to expand during meiosis to more than 200 repeats; her son would then be affected. These molecular changes are readily seen by straightforward molecular studies (Fig. 4). Females may also be affected if expansion occurs, but a less severe phenotype exists than males.
|
|
Females with the FRAXA premutation may show POF. Schwartz and colleagues154 reported that fragile X carrier females more often showed oligomenorrhea than noncarrier female relatives (38% versus 6%). Murray and colleagues155 analyzed 1268 controls, 50 familial POF cases, and 244 sporadic POF cases. Of familial cases, 16% showed FRAXA premutation; among sporadic cases, only 1.6% showed POF (Table 7). In the same sample POF was not increased in FRAXE. An international collaborative survey156 of 395 premutation carriers revealed that 63 (16%) underwent menopause under 40 years of age; only 0.4% of controls did. Surprisingly, frequency of POF was not increased in 128 FRAXA cases having a full mutation. Consistent with the above are observations that heterozygous FRAXA women respond poorly to ovulation-inducing agents, producing fewer oocytes and fewer embryos in assisted reproductive technologies (ART).157
Table 7. Premature ovarian failure and fragile X
FRAXA Premutation | |
Sporadic POF | 2/122 (1.6%) |
Familial POF | 4/25 (16%) |
Control | 0/634 |
Data of Murray and colleagues.155
POF, premature ovarian failure.
Overall FRAXA is associated with POF.158 The likelihood of fragile X premutation is 1–5% with sporadic POF and 10–15% with familial POF.158
MYOTONIC DYSTROPHY AND EXPANSION OF TRIPLET REPEATS (CTG)
Myotonic dystrophy is an autosomal dominant disorder characterized by muscle wasting (head, neck, extremities), frontal balding, cataracts, and male hypogonadism (80%) caused by testicular atrophy. Female hypogonadism is much less common, if increased at all. Despite frequent textual citations, ovarian failure is actually not well documented.
Pathogenesis of myotonic dystrophy involves nucleotide expansion of CTG in the 3[prime] untranslated region of a protein located on chromosome 19. Normally, five to 27 CTG repeats are present. Heterozygotes usually have at least 50 repeats; severely affected individuals show 600 or more. As in FRAXA, poor response to ovulation induction regions is observed. Sermon and colleagues159 report fewer embryos per cycle than in standard ART; thus, pregnancy rates in preimplantation genetic diagnosis are decreased.
AIRE (AUTOIMUNE POLY-ENDOCRINOPATHY-CANDIDIASIS ECTODERMAL DYSTROPHY, APECED)
The AIRE (autoimmune regulation) gene is located on 21q22.3, and is responsible for APECED. Many different AIRE perturbations have been found in this autosomal dominant disorder, a pleiotropic condition160 of varied expressivity. These include nonsense mutations and frame shifts, so causality is not in doubt.
Ovarian hypoplasia exists in 50–60% of cases.161 Other findings include alopecia, vitiligo, keratopathy, malabsorption, hepatitis, and mucocutaneous candidiasis. The wide range of features makes it unclear when to apply the APECED appelation. This becomes relevant to isolated POF; AIRE thus may or may not be a good candidate gene. There has been no attempt to determine if a particular mutation leads to ovarian failure, as distinct from its association with other autoimmune phenomena of APECED.
More specific mutations could otherwise be sought in isolated POF. AIRE mutations have not yet been sought in POF cases lacking autoimmune findings.
SYMPHALANGISM AND NOGGIN (NOG)
Noggin is responsible for the autosomal dominant disorder proximal symphalangism (SYM1).162 Characteristic features include ankylosis of the PIP (proximal interphalangeal) joints, carpal-tarsal fusions, brachydactyly, and deafness. NOG is expressed in the ovary and is an antagonist of bone morphogenic protein 4 and 7, a member of a family of genes (e.g., BMP15) already considered as a candidate for POF.163
One SYM1 case has shown POF.164 As predicted, a NOG mutation was found. Apparently NOG perturbations have not been sought in isolated POF subjects.
Premature ovarian failure and polyglandular autoimmune syndrome
Type 1 polyglandular autoimmune syndrome is an autosomal recessive disorder characterized by deficiencies of the parathyroids, adrenals and gonad exist; moniliasis secondary to immune deficiencies is common. Hypergonadotropic ovarian failure occurs in 60% of cases. The disorder is common in Finland and in the Iranian Jewish population,165 but rare in other populations. Localized to human 2q22.3 the gene has a murine homologue: autoimmune regulation gene (AIRE).
Type II polyglandular autoimmune syndrome is also called Schmidt syndrome. Inheritance is autosomal dominant. Failure or hypofunction occurs in gonads, adrenals, thyroid, and pancreas. Hematologic, gastrointestinal, ocular, and integumental (hair) defects also occur; immunologic dysfunction is often pronounced.
Anti-ovarian antibodies and oophoritis
Anti-ovarian antibodies may be either generalized in nature or directed against a specific cellular component (e.g., gonadotropin receptor, stromal cells, zona pellucida). Anasti166 reviewed the role of anti-ovarian antibodies in ovarian failure. In our opinion, a casual relation seems less likely than a secondary effect (epiphenomenon), with antibodies arising only after ovarian damage has occurred for unrelated but primary reasons.
Similar reasoning applies to oophoritis as a common cause of premature ovarian failure.
Mitochondrial genes
Perturbations of mitochondrial DNA (mtDNA) cause various disorders, usually involving muscle and neurologic systems. Searching for perturbations of mitochondrial DNA in POF should be fruitful because the mature oocyte contains the greatest number of mDNA copies of any human cell. The prototype is polymerase gamma progressive external ophthalmoplegia (PEO).
Proximal myopathy, sensory ataxia, and parkinsonism occur in this disorder. Among seven families studied by Luoma et al.167 POF co-segregated with PEO in three of them. The mutation Y955C was found in two of the three families. The tyrosine to cystosine change involves a highly conserved region, making a functional effect highly plausible. In the third family compound heterozygosity (N468D/A1105T) was observed in an affected woman.
REFERENCES
Smith A, Fraser IS, Noel M: Three siblings with premature gonadal failure. Fertil Steril 32:528, 1979 |
|
Granat M, Amar A, Mor-Yosef S, et al: Familial gonadal germinative failure: Endocrine and human leukocyte antigen studies. Fertil Steril 40:215, 1983 |
|
Hamet P, Kuchel O, Nowaczynski W, et al: Hypertension with adrenal, genital, renal defects, and deafness. A new familial syndrome Arch Intern Med 131:563, 1973 |
|
Al Awadi SA, Farag TI, Teebi AS, et al: Primary hypogonadism and partial alopecia in three sibs with mullerian hypoplasia in the affected females. Am J Med Genet 22:619, 1985 |
|
Mikati MA, Najjar SS, Sahli IF, et al: Microcephaly, hypergonadotropic hypogonadism, short stature, and minor anomalies: a new syndrome. Am J Med Genet 22:599, 1985 |
|
Simpson JL: Gonadal dysgenesis and sex chromosome abnormalities. Phenotypic/karyotypic correlations In Vallet HL, Perter IH (eds): Genetic Mechanisms of Sexual Development. pp 365, 405 New York, Academic Press, 1994 |
|
Meyers CM, Boughman JA, Rivas M, et al: Gonadal (ovarian) dysgenesis in 46,XX individuals: frequency of the autosomal recessive form. Am J Med Genet 63:518, 1996 |
|
Simpson JL, Christakos AC, Horwith M, et al: Gonadal dysgenesis associated with apparently normal chromosomal complements. Birth Defects 12(6):215, 1971 |
|
Boczkowski K: Pure gonadal dysgenesis and ovarian dysplasia in sisters. Am J Obstet Gynecol 106:626, 1970 |
|
Aittomaki K: The genetics of XX gonadal dysgenesis. Am J Hum Genet 54:844, 1994 |
|
Portuondo JA, Neyro JL, Benito JA, et al: Familial 46,XX gonadal dysgenesis. Int J Fertil 32:56, 1987 |
|
Aittomaki K, Lucena JL, Pakarinen P, et al: Mutation in the follicle-stimulating hormone receptor gene causes hereditary hypergonadotropic ovarian failure. Cell 82:959, 1995 |
|
Luoh SW, Bain PA, Polakiewicz RD et al: Zfx mutation results in small animal size and reduced germ cell number in male and female mice. Development 124:2275,1997 |
|
Pellas TC, Ramachandran B, Duncan M, et al: Germ-cell deficient (gcd), an insertional mutation manifested as infertility in transgenic mice. Proc Natl Acad Sci USA 88:8787, 1991 |
|
Manova K, Bachvarova RF: Expression of c-kit encoded at the W locus of mice in developing embryonic germ cells and presumptive melanoblasts. Dev Biol 146:312, 1991 |
|
Matsui Y, Zsebo KM, Hogan BL: Embryonic expression of a haematopoietic growth factor encoded by the Sl locus and the ligand for c-kit. Nature 347:667, 1990 |
|
Luo X, Ikeda Y, Parker KL: A cell-specific nuclear receptor is essential for adrenal and gonadal development and sexual differentiation. Cell 77:481, 1994 |
|
Edelmann W, Cohen PE, Kneitz B, et al: Mammalian MutS homologue 5 is required for chromosome pairing in meiosis. Nat Genet 21:123, 1999 |
|
Ratts VS, Flaws JA, Kolp R, et al: Ablation of bcl-2 gene expression decreases the numbers of oocytes and primordial follicles established in the post-natal female mouse gonad. Endocrinology 136:3665, 1995 |
|
Soyal S, Rankin TDJ: International Workshop of Early Folliculogenesis and Oocytes Development: basis and clinical aspects. London: Abstract Presentation 1999 |
|
Dong J, Albertini DF, Nishimori K, et al: Growth differentiation factor-9 is required during early ovarian folliculogenesis. Nature 383:531, 1996 |
|
Kumar TR, Wang Y, Lu N, et al: Follicle stimulating hormone is required for ovarian follicle maturation but not male fertility. Nat Genet 15:201, 1997 |
|
Dierich A, Sairam MR, Monaco L, et al: Impairing follicle-stimulating hormone (FSH) signaling in vivo: targeted disruption of the FSH receptor leads to aberrant gametogenesis and hormonal imbalance. Proc Natl Acad Sci USA 95:13612, 1998 |
|
Lubahn DB, Moyer JS, Golding TS, et al: Alteration of reproductive function but not prenatal sexual development after insertional disruption of the mouse estrogen receptor gene. Proc Natl Acad Sci USA 90:11162, 1993 |
|
Simon AM, Goodenough DA, Li E, et al: Female infertility in mice lacking connexin 37. Nature 385:525, 1997 |
|
Edelmann W, Cohen PE, Kane M, et al: Meiotic pachytene arrest in MLH1-deficient mice. Cell 85:1125, 1996 |
|
Rankin T, Familari M, Lee E, et al: Mice homozygous for an insertional mutation in the Zp3 gene lack a zona pellucida and are infertile. Development 122:2903, 1996 |
|
Topilko P, Schneider-Maunoury S, Levi G, et al: Multiple pituitary and ovarian defects in Krox-24 (NGFI-A, Egr-1)-targeted mice. Mol Endocrinol 12:107, 1998 |
|
Perrault M, Klotz P, Housset E: Deux cas de syndrome de Turner avec surdi-mutite dans une meme fratrie. Bull Soc Med Hop Paris 67:79, 1951 |
|
Christakos AC, Simpson JL, Younger JB, et al: Gonadal dysgenesis as an autosomal recessive condition. Am J Obstet Gynecol 104:1027, 1969 |
|
Pallister PD, Opitz JM: The Perrault syndrome: Autosomal recessive ovarian dysgenesis with facultative, non-sex–limited sensorineural deafness. Am J Med Genet 4:239, 1979 |
|
McCarthy DJ, Opitz JM: Perrault syndrome in sisters. Am J Med Genet 22:629, 1985 |
|
Nishi Y, Hamamoto K, Kajiyama M, et al: The Perrault syndrome: Clinical report and review. Am J Med Genet 31:623, 1988 |
|
Matthews CH, Borgato S, Beck-Peccoz P et al: Primary amenorrhoea and infertility due to a mutation in the beta-subunit of Nat Genet. 1993 Sep;5(1):83-6. |
|
Layman LC, Lee EJ, Peak DB et al: Delayed puberty and hypogonadism caused by mutations in the follicle-stimulating N Engl J Med. 1997 Aug 28;337(9):607-11. |
|
Aittomaki K, Herva R, Stenman UH, et al: Clinical features of primary ovarian failure caused by a point mutation in the follicle-stimulating hormone receptor gene. J Clin Endocrinol Metab 81:3722, 1996 |
|
Layman LC, Amde S, Cohen DP, et al: The Finnish follicle-stimulating hormone receptor gene mutation is rare in North American women with 46,XX ovarian failure. Fertil Steril 69:300, 1998 |
|
Simoni M, Gromoll J, Nieschlag E: The follicle-stimulating hormone receptor: biochemistry, molecular biology, physiology, and pathophysiology. Endocr Rev 18:739, 1997 |
|
da Fonte Kohek MB, Batista MC, Russell et al: No evidence of the inactivating mutation (C566T) in the follicle-stimulating hormone receptor gene in Brazilian women with premature ovarian failure. Fertil Steril 70:565, 1998 |
|
de la Chesnaye E, Canto P, Ulloa-Aguirre A, et al: No evidence of mutations in the follicle-stimulating hormone receptor gene in Mexican women with 46,XX pure gonadal dysgenesis. Am J Med Genet 98:125, 2001 |
|
Liu JY, Gromoll J, Cedars MI, et al: Identification of allelic variants in the follicle-stimulating hormone receptor genes of females with or without hypergonadotropic amenorrhea. Fertil Steril 70:326, 1998 |
|
Simpson JL, Rajkovic A: Ovarian differentiation and gonadal failure. Am J Med Genet 89:186, 1999 |
|
Tapanainen JS, Aittomaki K, Min J, et al: Men homozygous for an inactivating mutation of the follicle-stimulating hormone (FSH) receptor gene present variable suppression of spermatogenesis and fertility. Nat Genet 15:205, 1997 |
|
Lofrano-Porto A, Barra GB, Giacomini LA et al: Luteinizing hormone beta mutation and hypogonadism in men and women. N Engl J Med. 2007 Aug 30;357(9):897-904. |
|
Latronico AC, Anasti J, Arnhold IJ, et al: Brief report: testicular and ovarian resistance to luteinizing hormone caused by inactivating mutations of the luteinizing hormone-receptor gene. N Engl J Med 334:507, 1996 |
|
Toledo SP, Brunner HG, Kraaij R, et al: An inactivating mutation of the luteinizing hormone receptor causes amenorrhea in a 46,XX female. J Clin Endocrinol Metab 81:3850, 1996 |
|
Cramer DW, Xu H, Harlow BL: Family history as a predictor of early menopause. Fertil Steril 64:740, 1995 |
|
Laue L, Wu SM, Kudo M, et al: A nonsense mutation of the human luteinizing hormone receptor gene in Leydig cell hypoplasia. Hum Mol Genet 4:1429, 1995 |
|
Skre H, Bassoe HH, Berg K, et al: Cerebellar ataxia and hypergonadotropic hypogonadism in two kindreds. Chance concurrence, pleiotropism or linkage? Clin Genet 9:234, 1976 |
|
De Michele G, Filla A, Striano S, et al: Heterogeneous findings in four cases of cerebellar ataxia associated with hypogonadism (Holmes’ type ataxia). Clin Neurol Neurosurg 95:23, 1993 |
|
Linssen WH, Van den Bent MJ, Brunner HG, et al: Deafness, sensory neuropathy, and ovarian dysgenesis: A new syndrome or a broader spectrum of Perrault syndrome? Am J Med Genet 51:81, 1994 |
|
Gottschalk ME, Coker SB, Fox LA: Neurologic anomalies of Perrault syndrome. Am J Med Genet 65:274, 1996 |
|
Fryns JP, Van Lingen C, Devriendt K, et al: Two adult females with a distinct familial mental retardation syndrome: non-progressive neurological symptoms with ataxia and hypotonia, similar facial appearance, hypergonadotrophic hypogonadism, and retinal dystrophy. J Med Genet 35:333, 1998 |
|
Amor DJ, Delatycki MB, Gardner RJ, et al: New variant of familial cerebellar ataxia with hypergonadotropic hypogonadism and sensorineural deafness. Am J Med Genet 99:29, 2001 |
|
Maximilian C, Ionescu B, Bucur A: [Two sisters with major gonadal dysgenesis, dwarfism, microcephaly, arachnodactyly,and normal karyotype 46,XX]. J Genet Hum 18:365, 1970 |
|
Malouf J, Alam S, Kanj H, et al: Hypergonadotropic hypogonadism with congestive cardiomyopathy: Anautosomal-recessive disorder? Am J Med Genet 20:483, 1985 |
|
Quayle SA, Copeland KC: 46,XX gonadal dysgenesis with epibulbar dermoid. Am J Med Genet 40:75, 1991 |
|
Pober BR, Zemel S, Hisama FM: 46,XX gonadal dysgenesis, short stature and recurrent metabolic acidosis in two sisters. Am J Hum Genet 63:A117, 1998 |
|
Hisama FM, Zemel S, Cherniske EM, et al: 46,XX gonadal dysgenesis, short stature, and recurrent metabolic acidosis in two sisters. Am J Med Genet 98:121, 2001 |
|
Zlotogora J, Sagi M, Cohen T: The blepharophimosis, ptosis, and epicanthus inversus syndrome: Delineation of two types. Am J Hum Genet 35:1020, 1983 |
|
Crisponi L, Deiana M, Loi A, et al: The putative forkhead transcription factor FOXL2 is mutated in blepharophimosis/ptosis/epicanthus inversus syndrome. Nat Genet 27:159, 2001 |
|
Bailey WA, Zwingman TA, Reznik VM, et al: End-stage renal disease and primary hypogonadism associated with a 46,XX karyotype. Am J Dis Child 146:1218, 1992 |
|
Narahara K, Kamada M, Takahashi Y, et al: Case of ovarian dysgenesis and dilated cardiomyopathy supports existence of Malouf syndrome. Am J Med Genet 44:369, 1992 |
|
Guazzarotti L, Caprio C, Rinne TK et al: Limb-mammary syndrome (LMS) associated with internal female genitalia dysgenesia:a new genotype/phenotype correlation? Am J Med Genet A. 2008 Aug 1;146A(15):2001-4. |
|
Zadik Z, Levin S, Prager-Lewin R, Laron Z: Gonadal dysfunction in patients with ataxia telangiectasia. Acta Paediatr Scand. 4: 477, 1978 |
|
Waldmann TA, Misiti J, Nelson DL, et al: Ataxia-telangiectasis: A multisystem hereditary disease with immunodeficiency, impaired organ maturation, x-ray hypersensitivity, and a high incidence of neoplasia. Ann Intern Med 99:367, 1983 |
|
Matthijs G, Schollen E, Pardon E, et al: Mutations in PMM2, a phosphomannomutase gene on chromosome 16p13, in carbohydrate-deficient glycoprotein type I syndrome (Jaeken syndrome). Nat Genet 16:88, 1997 |
|
Kristiansson B, Stibler H, Wide L: Gonadal function and glycoprotein hormones in the carbohydrate-deficient glycoprotein (CDG) syndrome. Acta Paediatr 84:655, 1995 |
|
Nance MA, Berry SA: Cockayne syndrome: Review of 140 cases. Am J Med Genet 42:68, 1992 |
|
Sugarman GI, Landing BH, Reed WB: Cockayne syndrome: clinical study of two patients and neuropathologic findings in one. Clin Pediatr 16:225, 1977 |
|
Kaufman FR, Kogut MD, Donnell GN, et al: Hypergonadotropic hypogonadism in female patients with galactosemia. N Engl J Med 304:994, 1981 |
|
Waggoner DD, Buist NR, Donnell GN: Long-term prognosis in galactosaemia: results of a survey of 350 cases. J Inherit Metab Dis 13:802, 1990 |
|
Levy HL, Driscoll SG, Porensky RS, et al: Ovarian failure in galactosemia. N Engl J Med 310:50, 1984 |
|
Martsolf JT, Hunter AG, Haworth JC: Severe mental retardation, cataracts, short stature, and primary hypogonadism in two brothers. Am J Med Genet 1:291, 1978 |
|
Harbord MG, Baraitser M, Wilson J: Microcephaly, mental retardation, cataracts, and hypogonadism in sibs: Martsolf’s syndrome. J Med Genet 26:397, 1989 |
|
Hennekam RC, van de Meeberg AG, van Doorne JM, et al: Martsolf syndrome in a brother and sister: clinical features and pattern of inheritance. Eur J Pediatr 147:539, 1988 |
|
Weemaes CM, Hustinx TW, Scheres JM, et al: A new chromosomal instability disorder: the Nijmegen breakage syndrome. Acta Paediatr Scand 70:557, 1981 |
|
Conley ME, Spinner NB, Emanuel BS, et al: A chromosomal breakage syndrome with profound immunodeficiency. Blood 67:1251, 1986 |
|
Chrzanowska KH, Kleijer WJ, Krajewska-Walasek M, et al: Eleven Polish patients with microcephaly, immunodeficiency, and chromosomal instability: the Nijmegen breakage syndrome. Am J Med Genet 57:462, 1995 |
|
Hall JG, Pallister PD, Clarren SK, et al: Congenital hypothalamic hamartoblastoma, hypopituitarism, imperforate anus and postaxial polydactyly: A new syndrome? Part I: Clinical, causal, and pathogenetic considerations Am J Med Genet 7:47, 1980 |
|
Starr DG, McClure JP, Connor JM: Non-dermatological complications and genetic aspects of the Rothmund-Thomson syndrome. Clin Genet 27:102, 1985 |
|
Goto M, Tanimoto K, Horiuchi Y, et al: Family analysis of Werner’s syndrome: A survey of 42 Japanese families with a review of the literature. Clin Genet 19:8, 1981 |
|
Cramer DW, Harlow BL, Barbieri RL, et al: Galactose-1-phosphate uridyl transferase activity associated with age at menopause and reproductive history. Fertil Steril 51:609, 1989 |
|
Kaufman FR, Devgan S, Donnell GN: Results of a survey of carrier women for the galactosemia gene. Fertil Steril 60:727, 728 1993 |
|
Leslie ND, Yager K, Bai S: A mouse model for transferase deficiency galactosemia. Am J Hum Genet 57:191, 1995 |
|
Bjursell C, Stibler H, Wahlstrom J, et al: Fine mapping of the gene for carbohydrate-deficient glycoprotein syndrome, type I (CDG1): linkage disequilibrium and founder effect in Scandinavian families. Genomics 39:247, 1997 |
|
de Zegher F, Jaeken J: Endocrinology of the carbohydrate-deficient glycoprotein syndrome type 1 from birth through adolescence. Pediatr Res 37:395, 1995 |
|
Araki S, Chikazawa K, Sekiguchi I, et al: Arrest of follicular development in a patient with 17 alpha-hydroxylase deficiency: Folliculogenesis in association with a lack of estrogen synthesis in the ovaries. Fertil Steril 47:169, 1987 |
|
Rabinovici J, Blankstein J, Goldman B, et al: In vitro fertilization and primary embryonic cleavage are possible in 17 alpha-hydroxylase deficiency despite extremely low intrafollicular 17 beta-estradiol. J Clin Endocrinol Metab 68:693, 1989 |
|
Yanase T: 17alpha-Hydroxylase/17,20-lyase defects. J Steroid Biochem Mol Biol 53:153, 1995 |
|
Imai T, Yanase T, Waterman MR, et al: Canadian Mennonites and individuals residing in the Friesland region of The Netherlands share the same molecular basis of 17 alpha-hydroxylase deficiency. Hum Genet 89:95, 1992 |
|
Miura K, Yasuda K, Yanase T, et al: Mutation of cytochrome P-45017 alpha gene (CYP17) in a Japanese patient previously reported as having glucocorticoid-responsive hyperaldosteronism: With a review of Japanese patients with mutations of CYP17. J Clin Endocrinol Metab 81:3797, 1996 |
|
Kaneko S, Oshio S, Kobayashi T, et al: Human X- and Y-bearing sperm differ in cell surface sialic acid content. Biochem Biophys Res Commun 124:950, 1984 |
|
Simpson ER: Genetic mutations resulting in estrogen insufficiency in the male. Mol Cell Endocrinol 145:55, 1998 |
|
Ito Y, Fisher CR, Conte FA, et al: Molecular basis of aromatase deficiency in an adult female with sexual infantilism and polycystic ovaries. Proc Natl Acad Sci USA 90:11673, 1993 |
|
Conte FA, Grumbach MM, Ito Y, et al: A syndrome of female pseudohermaphrodism, hypergonadotropic hypogonadism, and multicystic ovaries associated with missense mutations in the gene encoding aromatase (P450arom). J Clin Endocrinol Metab 78:1287, 1994 |
|
Mullis PE, Yoshimura N, Kuhlmann B, et al: Aromatase deficiency in a female who is compound heterozygote for two new point mutations in the P450arom gene: Impact of estrogens on hypergonadotropic hypogonadism, multicystic ovaries, and bone densitometry in childhood. J Clin Endocrinol Metab 82:1739, 1997 |
|
Duck SC, Sekhon GS, Wilbois R, et al: Pseudohermaphroditis, with testes and a 46,XX karyotype. J Pediatr 87:58, 1975 |
|
Levinson G, Zarate A, Guzman-Toledano R, et al: An XX female with sexual infantilism, absent gonads, and lack of Mullerian ducts. J Med Genet 13:68, 1976 |
|
Mendonca BB, Barbosa AS, Arnhold IJ, et al: Gonadal agenesis in XX and XY sisters: Evidence for the involvement of an autosomal gene. Am J Med Genet 52:39, 1994 |
|
Kennerknecht I, Sorgo W, Oberhoffer R, et al: Familial occurrence of agonadism and multiple internal malformations in phenotypically normal girls with 46,XY and 46,XX karyotypes, respectively: A new autosomal recessive syndrome. Am J Med Genet 47:1166, 1993 |
|
Matthijs G, Schollen E, Pardon E et al: Mutations in PMM2, a phosphomannomutase gene on chromosome 16p13, in Nat Genet. 1997 May;16(1):88-92. |
|
Suzumori N, Pangas SA, Rajkovic A: Candidate genes for premature ovarian failure. Curr Med Chem. 2007;14(3):353-7. |
|
Conte FA, Grumbach MM, Ito Y et al: A syndrome of female pseudohermaphrodism, hypergonadotropic hypogonadism, and multicystic ovaries associated with missense mutations in the gene encoding aromatase (P450arom). J Clin Endocrinol Metab. 1994 Jun;78(6):1287-92. |
|
Dixit H, Deendayal M, Singh L: Mutational analysis of the mature peptide region of inhibin genes in Indian women with ovarian failure. Hum Reprod. 2004 Aug;19(8):1760-4. Epub 2004 Jun 17. |
|
Torgerson DJ, Thomas RE, Reid DM: Mothers and daughters menopausal ages: Is there a link? Eur J Obstet Gynecol Reprod Biol 74:63, 1997 |
|
Treloar SA, Do KA, Martin NG: Genetic influences on the age at menopause. Lancet 352:1084, 1998 |
|
Snieder H, MacGregor AJ, Spector TD: Genes control the cessation of a woman’s reproductive life: A twin study of hysterectomy and age at menopause. J Clin Endocrinol Metab 83:1875, 1998 |
|
Treloar SA, Martin NG, Heath AC: Longitudinal genetic analysis of menstrual flow, pain, and limitation in a sample of Australian twins. Behav Genet 28:107, 1998 |
|
Guttenbach M, Engel W, Schmid M: Analysis of structural and numerical chromosome abnormalities in sperm of normal men and carriers of constitutional chromosome aberrations. A review Hum Genet 100:1, 1997 |
|
Gekas J, Thepot F, Turleau C, et al: Chromosomal factors of infertility in candidate couples for ICSI: an equal risk of constitutional aberrations in women and men. Hum Reprod 16:82, 2001 |
|
Kim TJ, Anasti JN, Flack MR, et al: Routine endocrine screening for patients with karyotypically normal spontaneous premature ovarian failure. Obstet Gynecol 89:777, 1997 |
|
Karlsson FA, Kampe O, Winqvist O, et al: Autoimmune disease of the adrenal cortex, pituitary, parathyroid glands and gastric mucosa. J Intern Med 234:379, 1993 |
|
LaBarbera AR, Miller MM, Ober C, et al: Autoimmune etiology in premature ovarian failure. Am J Reprod Immunol Microbiol 16:115, 1988 |
|
Rebar RW, Erickson GF, Yen SS: Idiopathic premature ovarian failure: Clinical and endocrine characteristics. Fertil Steril 37:35, 1982 |
|
Alper MM, Garner PR: Premature ovarian failure: Its relationship to autoimmune disease. Obstet Gynecol 66:27, 1985 |
|
Belvisi L, Bombelli F, Sironi L, et al: Organ-specific autoimmunity in patients with premature ovarian failure. J Endocrinol Invest 16:889, 1993 |
|
Coulam CB, Stringfellow S, Hoefnagel D: Evidence for a genetic factor in the etiology of premature ovarian failure. Fertil Steril 40:693, 1983 |
|
Starup J, Sele V: Premature ovarian failure. Acta Obstet Gynecol Scand 52:259, 1973 |
|
Austin GE, Coulam CB, Ryan RJ: A search for antibodies to luteinizing hormone receptors in premature ovarian failure. Mayo Clin Proc 54:394, 1979 |
|
Mattison DR, Evans MI, Schwimmer WB, et al: Familial premature ovarian failure. Am J Hum Genet 36:1341, 1984 |
|
Testa G, Vegetti W, Tibiletti MC, et al: Pattern of inheritance in familial premature ovarian failure. Hum Reprod 12:P-174, 1997 |
|
Ragni G, Parazzini F, Sapienza F, et al: Semen parameters and conception rates after intraperitoneal insemination. Gynecol Obstet Invest 44:239, 1997 |
|
Vegetti W, Marozzi A, Manfredini E, et al: Premature ovarian failure. Mol Cell Endocrinol 161:537, 2000 |
|
Vegetti W, Grazia TM, Testa G, et al: Inheritance in idiopathic premature ovarian failure: Analysis of 71 cases. Hum Reprod 13:1796, 1998 |
|
Shelling AN, Burton KA, Chand AL et al: Inhibin: a candidate gene for premature ovarian failure. Hum Reprod. 2000 Dec;15(12):2644-9. |
|
Marozzi A, Porta C, Vegetti W et al: Mutation analysis of the inhibin alpha gene in a cohort of Italian women affected by ovarian failure. Hum Reprod. 2002 Jul;17(7):1741-5. |
|
Araki S, Chikazawa K, Sekiguchi I et al: Arrest of follicular development in a patient with 17 alpha-hydroxylase deficiency: folliculogenesis in association with a lack of estrogen synthesis in the ovaries. Fertil Steril. 1987 Jan;47(1):169-72. |
|
Suzumori N, Yan C, Matzuk MM et al: Nobox is a homeobox-encoding gene preferentially expressed in primordial and growing oocytes. Mech Dev. 2002 Feb;111(1-2):137-41. |
|
Qin Y, Choi Y, Zhao H et al: NOBOX homeobox mutation causes premature ovarian failure. Am J Hum Genet. 2007 Sep;81(3):576-81. Epub 2007 Jul 10. |
|
Pangas SA, Choi Y, Ballow DJ et al: Oogenesis requires germ cell-specific transcriptional regulators Sohlh1 and Lhx8. Proc Natl Acad Sci U S A. 2006 May 23;103(21):8090-5. Epub 2006 May 11. |
|
Qin Y, Zhao H, Kovanci E et al: Analysis of LHX8 mutation in premature ovarian failure. Fertil Steril. 2008 Apr;89(4):1012-4. Epub 2007 Jul 10. |
|
Tsuda M, Sasaoka Y, Kiso M et al: Conserved role of nanos proteins in germ cell development. Science. 2003 Aug 29;301(5637):1239-41. |
|
Dixit H, Rao LK, Padmalatha V et al: Mutational screening of the coding region of growth differentiation factor 9 gene in Indian women with ovarian failure. Menopause. 2005 Nov-Dec;12(6):749-54 |
|
Dong J, Albertini DF, Nishimori K et al: Growth differentiation factor-9 is required during early ovarian folliculogenesis. Nature. 1996 Oct 10;383(6600):531-5. |
|
Kovanci E, Rohozinski J, Simpson JL et al: Growth differentiating factor-9 mutations may be associated with premature ovarian failure. Fertil Steril. 2007 Jan;87(1):143-6. Epub 2006 Dec 6. |
|
Zhao H, Qin Y, Kovanci E et al: Analyses of GDF9 mutation in 100 Chinese women with premature ovarian failure. Fertil Steril. 2007 Nov;88(5):1474-6. Epub 2007 May 7. |
|
Kovanci E, Simpson JL, Amato P et al: Oocyte-specific G-protein-coupled receptor 3 (GPR3): no perturbations found in 82women with premature ovarian failure (first report). Fertil Steril. 2007 Oct 20. |
|
Katsuya T, Horiuchi M, Minami S et al: Genomic organization and polymorphism of human angiotensin II type 2 receptor: no evidence for its gene mutation in two families of human premature ovarian failure syndrome. Mol Cell Endocrinol. 1997 Mar 28;127(2):221-8. |
|
Richards KA, Fukai K, Oiso N et al: A novel KIT mutation results in piebaldism with progressive depigmentation. J Am Acad Dermatol. 2001 Feb;44(2):288-92. |
|
Shibanuma K, Tong ZB, Vanderhoof VH et al: Investigation of KIT gene mutations in women with 46,XX spontaneous premature ovarian failure. BMC Womens Health. 2002 Aug 2;2(1):8. |
|
Durlinger AL, Kramer P, Karels B et al: Control of primordial follicle recruitment by anti-Mullerian hormone in the mouse ovary. Endocrinology. 1999 Dec;140(12):5789-96. |
|
Wang HQ, Takakura K, Takebayashi K et al: Mutational analysis of the mullerian-inhibiting substance gene and its receptor gene in Japanese women with polycystic ovary syndrome and premature ovarian failure. Fertil Steril. 2002 Dec;78(6):1329-30. |
|
Suzumori N, Burns KH, Yan W et al: RFPL4 interacts with oocyte proteins of the ubiquitin-proteasome degradation pathway. Proc Natl Acad Sci U S A. 2003 Jan 21;100(2):550-5. Epub 2003 Jan 13. |
|
Zlotogora J, Sagi M, Cohen T: The blepharophimosis, ptosis, and epicanthus inversus syndrome: delineation oftwo types. Am J Hum Genet. 1983 Sep;35(5):1020-7. |
|
Crisponi L, Deiana M, Loi A et al: The putative forkhead transcription factor FOXL2 is mutated inblepharophimosis/ptosis/epicanthus inversus syndrome. Nat Genet. 2001 Feb;27(2):159-66. |
|
De Baere E, Dixon MJ, Small KW et al: Spectrum of FOXL2 gene mutations in blepharophimosis-ptosis-epicanthus inversus Hum Mol Genet. 2001 Jul 15;10(15):1591-600. |
|
Harris SE, Chand AL, Winship IM et al: Identification of novel mutations in FOXL2 associated with premature ovarian failure. Mol Hum Reprod. 2002 Aug;8(8):729-33 |
|
Watkins WJ, Umbers AJ, Woad KJ et al: Mutational screening of FOXO3A and FOXO1A in women with premature ovarian failure. Fertil Steril. 2006 Nov;86(5):1518-21 |
|
Panidis D, Rousso D, Vavilis D, et al: Familial blepharophimosis with ovarian dysfunction. Hum Reprod 9:2034, 1994 |
|
Fraser IS, Shearman RP, Smith A, et al: An association among blepharophimosis, resistant ovary syndrome, and true premature menopause. Fertil Steril 50:747, 1988 |
|
Harrar HS, Jeffery S, Patton MA: Linkage analysis in blepharophimosis-ptosis syndrome confirms localisation to 3q21-24. J Med Genet 32:774, 1995 |
|
Amati P, Gasparini P, Zlotogora J, et al: A gene for premature ovarian failure associated with eyelid malformation maps to chromosome 3q22-q23. Am J Hum Genet 58:1089, 1996 |
|
Schwartz CE, Dean J, Howard-Peebles PN, et al: Obstetrical and gynecological complications in fragile X carriers: a multicenter study. Am J Med Genet 51:400, 1994 |
|
Murray A, Webb J, Grimley S, et al: Studies of FRAXA and FRAXE in women with premature ovarian failure. J Med Genet 35:637, 1998 |
|
Allingham-Hawkins DJ, Babul-Hirji R, Chitayat D, et al: Fragile X premutation is a significant risk factor for premature ovarian failure: The International Collaborative POF in Fragile X study. Preliminary data Am J Med Genet 83:322, 1999 |
|
Black SH, Levinson G, Harton GL, et al: Preimplantation genetic testing (PGT) from fragile X (fraxX). Am J Hum Genet 57:A31, 1995 |
|
Wittenberger MD, Hagerman RJ, Sherman SL et al: The FMR1 premutation and reproduction. Fertil Steril. 2007 Mar;87(3):456-65 |
|
Sermon K, De Vos A, Van de Velde V, et al: Fluorescent PCR and automated fragment analysis for the clinical application of preimplantation genetic diagnosis of myotonic dystrophy (Steinert’s disease). Mol Hum Reprod 4:791, 1998 |
|
Wang CY, Davoodi-Semiromi A, Huang W et al: Characterization of mutations in patients with autoimmune polyglandular syndrometype 1 (APS1). Hum Genet. 1998 Dec;103(6):681-5. |
|
Laml T, Preyer O, Umek W et al: Genetic disorders in premature ovarian failure. Hum Reprod Update. 2002 Sep-Oct;8(5):483-91. |
|
Perrault M, Klotz B, Housset E: [Two cases of Turner syndrome with deaf-mutism in two sisters.] Bull Mem Soc Med Hop Paris. 1951 Jan 26-Feb 2;67(3-4):79-84. |
|
Nishi Y, Hamamoto K, Kajiyama M et al: The Perrault syndrome: clinical report and review. Am J Med Genet. 1988 Nov;31(3):623-9. |
|
Pallister PD, Opitz JM: The Perrault syndrome: autosomal recessive ovarian dysgenesis with facultative, Am J Med Genet. 1979;4(3):239-46. |
|
Ahonen P, Myllarniemi S, Sipila I, et al: Clinical variation of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) in a series of 68 patients. N Engl J Med 322:1829, 1990 |
|
Anasti JN: Premature ovarian failure: An update. Fertil Steril 70:1, 1998 |
|
Luoma P, Melberg A, Rinne JO et al: Parkinsonism, premature menopause, and mitochondrial DNA polymerase gamma mutations: clinical and molecular genetic study. Lancet. 2004 Sep 4-10;364(9437):875-82. |