The Hypothalamic-Hypophyseal-Ovarian Axis and the Menstrual Cycle
Authors
INTRODUCTION
The menstrual cycle requires precise coordination between several processes in the body.1, 2 The major components of this control system include the hypothalamic gonadotropin-releasing hormone (GnRH) pulse generator, the pituitary gonadotropes, the ovaries, and the uterus. The gonadotropes respond to GnRH pulses by releasing the gonadotropins, follicle-stimulating hormone (FSH), and luteinizing hormone (LH), which stimulate folliculogenesis and steroid and peptidergic hormone secretion from the ovaries. Hypothalamic and pituitary activities are strictly controlled by ovarian hormone feedback loops, whereas the GnRH pulse generator is also modulated by a variety of inputs from other neural centers.
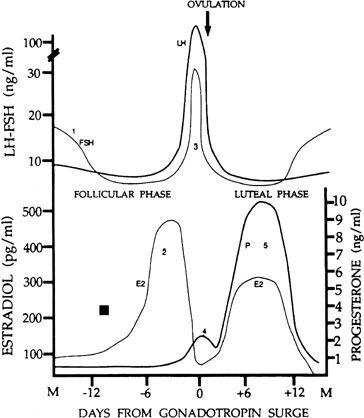
The menstrual cycle is divided into two successive phases. The follicular phase represents the process whereby a follicle is selected and becomes a mature follicle destined to ovulate. This first phase is dominated by estradiol secretion. Ovulation in response to a large release of gonadotropins signals the beginning of the second phase, the luteal phase, in which the ovulated follicle is transformed into a corpus luteum. The dominant ovarian hormone secreted in this phase is progesterone, supplemented by estradiol. Changing cyclic ovarian steroid hormone patterns prepare the uterus for implantation, were fertilization to occur in that cycle. At the end of the luteal phase, ovarian steroid hormone secretion collapses. This terminates the support of the endometrium, and menstruation occurs. The menstrual cycle lasts for 25–30 days in most women. By convention, the day of menstruation is designated as day 1 of the menstrual cycle, although the FSH signal initiating the cycle (see later) may occur 2–3 days before this. Although the follicular phase lasts 14 days in the typical 28-day cycle, in reality its length is variable. In contrast, the life span of the corpus luteum is remarkably constant, and the luteal phase lasts 13–15 days.
CLASSIC HORMONAL MARKERS OF THE CYCLE
There are four major circulating hormones that can easily be monitored during the menstrual cycle: FSH, LH, estradiol, and progesterone. Although the concentrations of these hormones in blood can vary substantially on an hourly basis, their daily profiles provide characteristic changes during the cycle (Fig. 1). The important event in regard to FSH is a small but significant rise in its levels at the end of the preceding cycle. This rise heralds the recruitment of secondary early antral follicles and the process whereby a follicle will be selected for ovulation. The most striking change in LH secretion occurs at the end of the follicular phase, when there is an abrupt rise in its concentration. This is the preovulatory gonadotropin surge that initiates the ovulatory process. Estradiol reflects the secretory activity of the growing follicle, and its concentrations rise in parallel with follicular growth, reaching highest levels when the follicle achieves maturation. Progesterone reflects the secretory activity of the corpus luteum. It rises and falls in a characteristic 14-day bell-shaped curve representative of the finite life span of the corpus luteum. Estradiol is also secreted by the corpus luteum in a similar rise-and-fall fashion. Other hormones also are released in a cyclic fashion (e.g., the inhibins) (see below).
THE HYPOTHALAMIC-HYPOPHYSEAL-OVARIAN AXIS
The hypothalamic unit and the gonadotropin-releasing hormone (GnRH) pulse generator
The GnRH pulse generator is the primary structure that drives the menstrual cycle. In the absence of a functional GnRH pulse generator, the gonadotropes remain unstimulated and the ovaries dormant. The GnRH pulse generator is the hypothalamic structure that releases GnRH, a decapeptide that stimulates LH and FSH synthesis and release from the pituitary. GnRH is synthesized in specialized neurons as part of a larger precursor molecule (prohormone) containing 92 amino acids that is processed before release during axonal transport from the hypothalamic neuron to the median eminence.3 Although GnRH neurons in the hypothalamus appear as a dispersed population spread over several classic architectonic areas,4 the primary GnRH neurons controlling the pituitary gland in primates appear to be located within the arcuate nucleus (a structure that contains several other small peptides, neurotransmitters, and endogenous opiates).5 Upon reaching the median eminence, GnRH axons end on capillary loops that give rise to long portal veins that descend along the pituitary stalk to terminate in anterior pituitary sinusoids. This anatomic arrangement is essential because it allows for the rapid and undiluted transport of the neurohormone, which has a half-life of only a few minutes, to its target receptor on the gonadotrope in the anterior pituitary gland.
GnRH neurons, in contrast to other neurons, do not originate within the brain.6 They derive from progenitor cells in the embryonic olfactory placode and during fetal development the GnRH neurons migrate into the brain to reach the locations that they will occupy during adult life.6 Functional connections between these neurons and the hypophyseal portal system are established by 16 weeks of fetal life.4 Completed migration failure and lack of establishment of functional connections are characteristic of patients with hypogonadotropic hypogonadism, which when accompanied by anosmia fullfills the diagnosis of Kallmann’s syndrome.7 In the 19-week fetus with X-linked Kallmann’s syndrome, the GnRH neurons accompanying the olfactory nerves are shown to be arrested in their voyage within the meninges, and contact with the brain and the hypophyseal portal system cannot be established.8 The migration of GnRH neurons over long distances and through changing molecular environments suggest that numerous factors, local and possibly external, influence this process at its different stages.9, 10 Factors that mediate the adhesion of GnRH neurons to changing surfaces along their voyage, that promote cytoskeletal remodeling and modulate axonal guidance, may play critical roles.11, 12
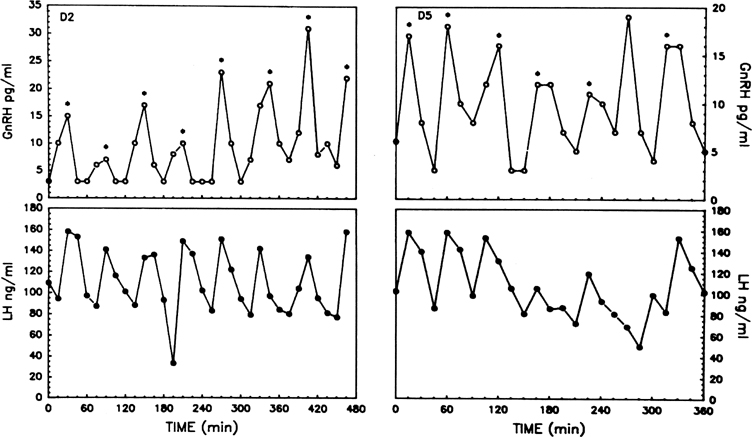
GnRH causes the release of both gonadotropins from the anterior pituitary gland. One essential aspect of gonadotropin secretion is that LH and FSH are released in a pulsatile rather than a continuous fashion. Each pulse of LH consists of the abrupt release of the hormone from the gonadotrope into the peripheral circulation, followed by an exponential decline representative of the half-life of the hormone. As is now well established in several animal species, pulsatile gonadotropin release is not the result of an intrinsic property of the anterior pituitary gland but is caused directly by the pulsatile release of GnRH (the GnRH pulse generator).5 Pulses of GnRH have been measured in hypophyseal portal blood and cerebrospinal fluid of nonhuman primates and sheep, and synchrony between pulses of GnRH in portal blood and of LH in peripheral blood is well shown.13, 14 Left to itself and in the absence of endogenous feedback systems and of exogenous influences, the pulse generator releases a GnRH pulse at about hourly intervals (Fig. 2). Pulsatility seems to be an intrinsic property of the GnRH neuron early on, because cultured neurons obtained from the olfactory placode and from migratory pathways in fetal life already release GnRH in a pulsatile manner.15 What accounts for the synchronous discharge of widely scattered GnRH neurons remains to be determined. One recently identified peptide, kisspeptin, may, among others, play a signaling role in this process: several studies have shown that kisspeptin stimulates the release of GnRH by activating receptors expressed by GnRH neurons.16
Fig. 2. Hourly pulsatile GnRH release during two periods of 8 hours in an ovariectomized monkey (upper levels). Note the concordance of LH (lower levels) and GnRH pulses. (Xia L, Van Vugt D, Alston EJ, et al: A surge of gonadotropin-releasing hormone accompanies the estradiol-induced gonadotropin surge in the rhesus monkey. Endocrinology 131: 2812, 1992; used by permission of The Endocrine Society.)
The hypothalamic GnRH pulse generator is operational at birth; significant pituitary and gonadal secretory activity occurs at that time and for a while thereafter. Later on, however, and throughout infancy until the prepubertal period, pulsatile gonadotropin secretion is significantly dampened.17 This is seen even in the absence of the gonads, as shown in patients with gonadal dysgenesis.18 Initiation of puberty is heralded by the resumption of a frank pulsatile pattern which is at first only seen in conjunction with deep sleep. This sleep-related pattern disappears in the later stages of puberty and is not apparent in adulthood when pulsatile secretion occurs throughout the 24-hour period. There are reports, however, of a nocturnal slowing of pulsatile LH release in a small number of women during the follicular phase of the menstrual cycle.19
The pituitary unit
The gonadotropins, LH and FSH, are synthesized within the gonadotrope in the anterior pituitary gland. These two hormones are glycoproteins containing two subunits (α and β). The β subunits of LH and FSH differ in their sequence. The β subunit confers hormone specificity. The α subunit is common to LH and FSH (as well as thyroid-stimulating hormone and human chorionic gonadotropin). Biologic activity of the hormone requires the association of both subunits.20
The relevance of pulsatile GnRH release to the reproductive process is illustrated by observations that only pulsatile GnRH administration, provided at an appropriate frequency, restores normal gonadotropin secretion in individuals lacking endogenous GnRH secretion.21 In contrast, continuous (nonpulsatile) exposure to GnRH, even to high doses, produces a desensitization of its receptor on the gonadotrope, resulting with time in a lowering of LH and FSH release.22, 23 The cellular mechanism of desensitization (also referred to as down-regulation) remains to be elucidated and may implicate receptor and postreceptor mechanisms.24
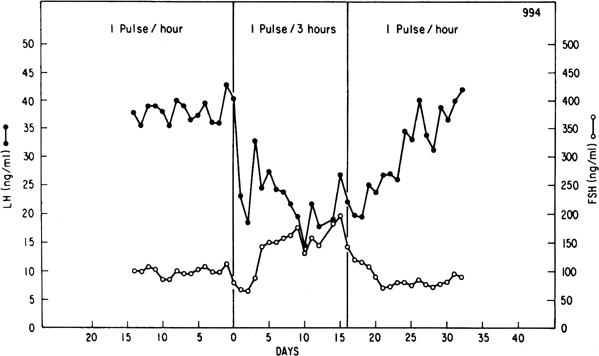
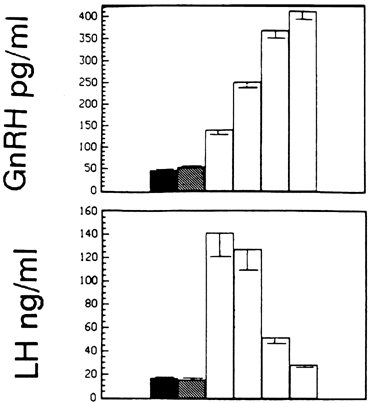
Although GnRH is known to release both LH and FSH, the relative amounts of each gonadotropin released may vary in response to ovarian feedback input (see later). In addition, it is now clear that not only is a pulsatile GnRH signal required for the stimulation of gonadotropin subunit gene transcription, but also that changes in the profile of pulsatile GnRH secretion differentially influence the synthesis and release of each gonadotropin. In particular, the frequency of GnRH pulse release selectively regulates gonadotropin subunit gene transcription with a faster frequency favoring LH β subunit gene expression and a lower frequency favoring FSH β subunit.25 In concurrence with this finding, studies in monkeys in which the GnRH pulse generator was lesioned and in GnRH-deficient patients show that a high GnRH pulse frequency infusion favors LH release, whereas a low GnRH pulse frequency favors FSH secretion26, 27, 28 (Fig. 3).
The ovarian unit and ovarian-hypothalamic and pituitary feedback loops
Both gonadotropins act on the ovaries to induce morphologic changes and ovarian steroid secretion. Morphologic processes include folliculogenesis (i.e., the cyclic recruitment of a pool of follicles to produce a mature follicle ready for ovulation) and the formation of a corpus luteum. These processes occur in sequence conferring a monthly rhythm to the reproductive cycle (see later). Granulosa and thecal cells within the follicle and luteal cells respond to LH by synthesizing and releasing ovarian steroids, mainly estradiol 17β and progesterone. The type and amount of hormone released depend on the status of the follicle and the corpus luteum.
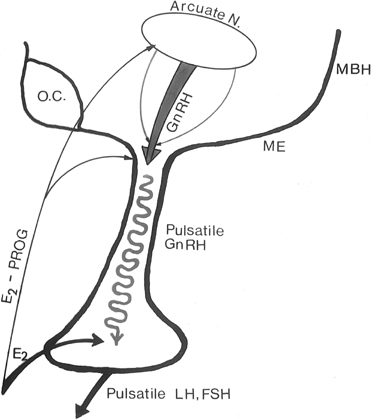
Feedback communication between the ovaries and the hypothalamic-pituitary unit is an essential component to the physiology of the reproductive cycle. It is important for the brain and pituitary gland to modulate their secretion in response to the activity status of the ovary. Estradiol and progesterone play a major role in these feedback communications. As in other endocrine systems, the major feedback loop is inhibitory (the negative feedback loop), whereby the steroid secreted by the target organ (the ovary) regulates the hypothalamic-hypophyseal unit to adjust GnRH and gonadotropin secretion (Fig. 4).
Fig. 4. In the primate, the gonadotropin-releasing hormone (GnRH) pulse generator is probably located within the arcuate nucleus of the hypothalamus. GnRH pulses are released in hypophyseal portal veins, which transport GnRH to the pituitary to stimulate pulsatile luteinizing hormone (LH) and follicle-stimulating hormone (FSH) release. These two gonadotropins stimulate folliculogenesis and steroid and peptidergic hormone secretion from the ovaries. Hypothalamic (GnRH) and pituitary (LH and FSH) activities are strictly controlled by ovarian hormone (estradiol and progesterone) feedback loops. MBH, mediobasal hypothalamus; ME, median eminence.
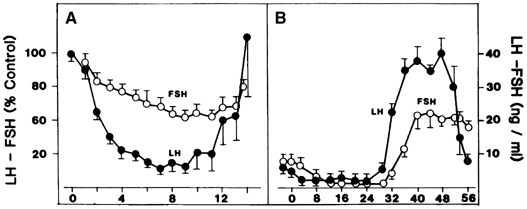
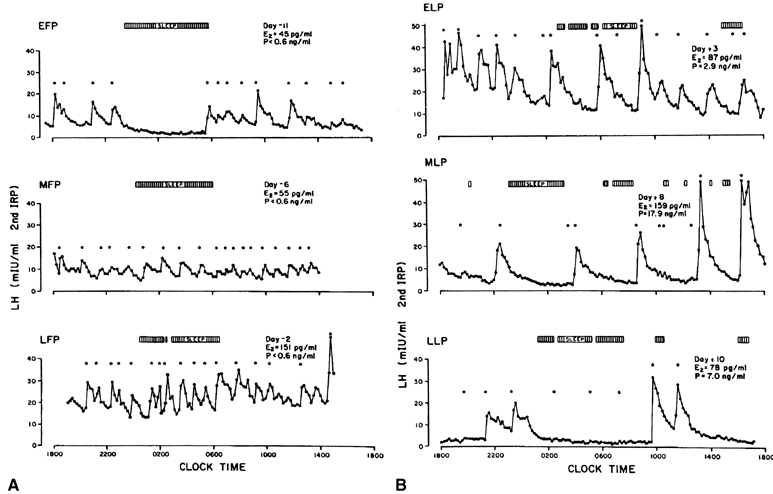
Estradiol-17β is a potent physiologic inhibitor of GnRH and of gonadotropin secretion.29 The threshold for the negative feedback action of estradiol is such that even small increases in the levels of the hormone induce a decrease in gonadotropins. Levels of LH and FSH during the follicular phase vary in accord with the changes in estradiol concentrations that accompany maturation of the follicle. As circulating estradiol levels increase during the follicular phase, gonadotropin concentrations decrease. In menopausal women or women who have undergone ovariectomy, in whom estradiol secretion is deficient, sustained increases in LH and FSH release occur. In these conditions, the administration of physiologic doses of estradiol results in a rapid and sustained decrease in LH and FSH to levels equivalent to those seen during the menstrual cycle (Fig. 5A).30, 31 The estradiol negative feedback loop acts to decrease LH secretion rapidly, mainly by controlling the amplitude of the LH pulse. As the follicular phase progresses, LH pulse amplitude declines. LH pulse frequency during the follicular phase (at 60–100-minute intervals) approximates that observed at menopause or after ovariectomy, suggesting that estradiol does not particularly affect LH pulse frequency. (At higher physiologic concentrations, estradiol also exerts a separate stimulatory [positive feedback loop] effect on gonadotropin secretion. This is described later.) Progesterone, at high concentrations such as those observed during the luteal phase of the cycle, also exerts an inhibitory effect on gonadotropin secretion. In contrast to estradiol, progesterone affects mainly LH pulse frequency. Experimentally a significant decrease in the frequency of the LH pulse can be induced by treating women in the follicular phase with progesterone.32
In view of these feedback loops, it is not surprising that the characteristics of pulsatile LH secretion vary greatly with the stage of the menstrual cycle (Fig. 6). During the estrogenic stage or follicular phase (Fig. 6A), pulses of high frequency but of low amplitude are seen, whereas during the progesterone stage or luteal phase (Fig. 6B), there is a progressive reduction in the frequency of the LH pulse, with pulse intervals occurring every 200 minutes or more by the end of the luteal phase. This decreased pulse frequency is accompanied by a significant increase in pulse amplitude.
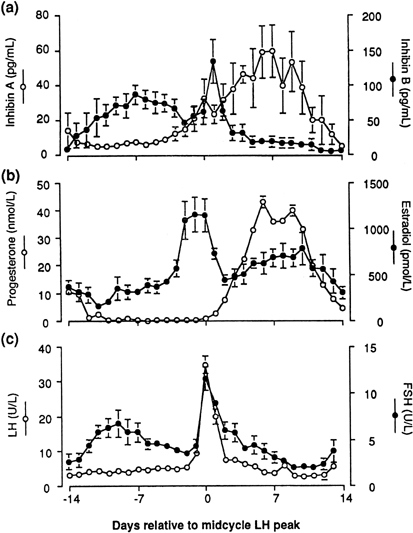
Inhibins, a family of peptides of ovarian origin, are glycoproteins that consist of a dimer, with two dissimilar α and β subunits.33 The two subunits are coded by different genes. Two forms of the β subunit have been identified and thus inhibin can exist as α-βA (inhibin A) and as α-βB (inhibin B). When measured throughout the menstrual cycle, plasma concentrations of inhibin B rise rapidly on the day after the intercycle FSH rise, remain elevated for a few days, then fall progressively during the remainder of the follicular phase. After a short-lived peak following the ovulatory gonadotropin surge, inhibin B falls to a low concentration during the luteal phase. In contrast, inhibin A concentrations rise only in the later part of the follicular phase and are maximal during the midluteal phase (Fig. 7).34 These different patterns of circulating inhibin B and inhibin A during the human menstrual cycle suggest different physiologic roles (see later).
Fig. 7. Mean plasma concentrations of inhibin A and inhibin B (upper panel) compared to estradiol and progesterone (center panel), and LH and FSH (lower panel) during the menstrual cycle. Day 0 is the day of the LH surge. (Groome NP, Illingworth PJ, O’Brien M et al: Measurement of dimeric inhibin B throughout the human menstrual cycle. J Clin Endocrinol Metab 81: 1401, 1996; used by permission of The Endocrine Society.) |
The inhibins influence FSH release specifically through their own negative feedback loop.35 This negative feedback loop, however, functions at a slower rate than that of the above-mentioned ovarian steroid negative feedback loop which is activated within minutes, and is directed mainly at the pituitary gland. It is believed that the decline in FSH after its peak in the early follicular phase of the normal cycle results from a negative feedback action of inhibin B at the pituitary level. This action has been shown only under experimental conditions, however, for example after antiestrogen therapy in the midfollicular phase.36 At menopause or in premature ovarian failure, data show a decreased secretion of inhibin with reproductive aging,37 suggesting that inhibin B negative feedback may be an important factor controlling the early monotropic increase in FSH with aging.38, 39 Other dimers of the β subunit, called activins, have also been reported to play a role in FSH release.35
THE MENSTRUAL CYCLE
The follicular phase
CYCLIC RECRUITMENT OF A COHORT
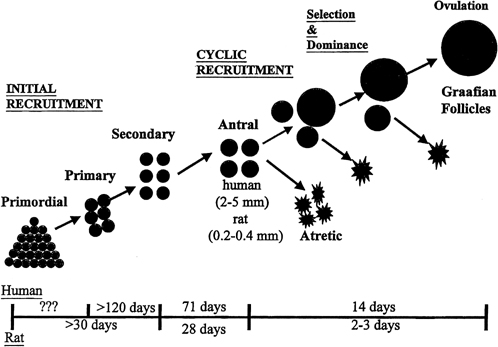
Although mechanisms of initial follicular growth in the ovary are difficult to investigate because of its protracted process, there is good evidence that dormant primordial follicles are recruited continuously into growing, a process that transforms them into primary, secondary, and early antral follicles, at which stage they become atretic. This process is initiated already before birth and continues until menopause and seems to proceed under the control of still unknown local ovarian factors, with gonadotropins perhaps playing a minor modulatory role.40 After puberty and in each menstrual cycle, one cohort of early antral follicles is rescued from going into the atretic process and recruited for further growth. This 'cyclic' antral follicle recruitment requires an appropriate signal from the pituitary gland (Fig. 8).41 This signal is FSH as represented by its small but selective increase starting at the end of the preceding luteal phase (see Fig. 1).42 The size of the cohort is dictated by the amount of FSH present at recruitment: a greater sized cohort can be recruited by increasing FSH amounts, as shown in gonadotropin-stimulated cycles in which hormones usually are administered at supraphysiologic levels.
FSH stimulates mitosis and activates aromatase production in granulosa cells, allowing for growth and increased local estradiol production. Data also show that FSH is the major stimulus for inhibin B secretion, which increases at that time.43 Experimental treatment with a GnRH antagonist at the end of the luteal phase, which abolishes the early follicular phase rise in FSH, also prevents the associated increase in inhibin B, whereas the same treatment followed by exogenous FSH restores the secretion of inhibin B.44 The measurement of circulating inhibin B levels in the early follicular phase or in the early days of a stimulated cycle provides an early indicator of the number of recruited follicles and of their activity.
SELECTION OF A DOMINANT FOLLICLE
Although several follicles are recruited in the early follicular phase as part of the cohort, in mono-ovular species usually only one continues to grow into a mature preovulatory follicle.45 This is referred to as the selection process, which occurs early in the midfollicular phase.46 Experimental cauterization of the largest follicle at that time in the monkey results in a delay in the midcycle gonadotropin surge, a reflection of the absence of any surrogate follicle able to immediately take the place of the destroyed selected follicle and of the need to start anew and recruit another cohort of follicles.
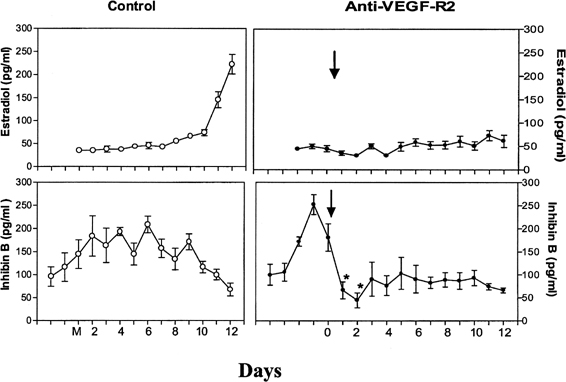
The precise mechanism by which one follicle of the cohort is selected in primates remains to be elucidated. The selected dominant follicle has a distinguishable structure, however, in terms of its cellular development and especially of its vascularization it possesses a denser microvascular network than that of lesser developed follicles.47 Experimental evidence has shown an active angiogenic process in this follicle destined for ovulation, suggesting an important role for regulators of angiogenesis in cyclic folliculogenesis. Vascular endothelial growth factor (VEGF), a major angiogenic factor, is detected in the developing follicle, with the most intense signal in the mature follicle.48 VEGF activity is well correlated with proliferating activity markers in vascular endothelial cells and is a sign of active vasculogenesis.49 Experimental data in nonhuman primates showed that VEGF receptor inactivation in the early follicular phase results in a rapid decrease in inhibin B secretion, suggesting an arrest in the development of the cohort of recruited antral follicles and in a delay in the rise in estradiol (Fig. 9).50 These data directly show that normal follicular development cannot occur in the absence of adequate VEGF stimulation to ensure proper local angiogenesis. Data in the literature also suggest a role for the gonadotropins in VEGF production by the follicle. Studies with monkey granulosa cells show that not only large amounts of gonadotropins representative of the midcycle ovulatory surge but also smaller amounts more typical of tonic secretion both enhance local VEGF production.51
GROWTH OF THE DOMINANT FOLLICLE
The morphologic hallmark of the selected follicle is the acquisition of LH receptors in response to FSH action52 and the differentiation of an endocrinologically active theca layer capable of synthesizing androgens in response to LH stimulation. These androgens are aromatized to estradiol in the granulosa cell layer, which contains aromatase but which itself does not possess the full complement of steroid biosynthetic enzymes required for the synthesis of androgens.53 Serum estradiol levels begin to rise as a result of the emergence of the dominant follicle. The rising estradiol, through the negative feedback loop, suppresses FSH levels (see Fig. 1) to concentrations that are too low to sustain maturation of the other follicles in the cohort with the consequence that these undergo final atresia.54 This process can be prevented experimentally by neutralizing the rising estradiol levels or by providing a moderate and continued elevation of FSH during the midfollicular phase in these instances, the physiologic process of single follicle dominance is interfered with, and ongoing growth of multiple follicles is fostered.55
By the midfollicular phase, there is an incremental increase in LH receptors, in aromatizable androgens, and in estrogens, which induces FSH receptors in the granulosa cells of the dominant follicle in a self-propagating mechanism. The developing dominant follicle generates its own estradiol microenvironment, which promotes further granulosa cell growth directly through its mitogenic activity or indirectly through the stimulation of local growth factors, such as insulin growth factor, members of the transforming growth factor superfamily (inhibins, activins), and others.56, 57
The pattern of pulsatile GnRH-driven LH secretion changes dynamically across the human menstrual cycle. During the follicular phase, a pulse frequency of about 1 pulse/90 minutes is the norm. Adherence to a specific regimen of pulse amplitude and frequency is crucial for a normal follicular phase, menstrual cyclicity, and reproductive function, and derangements of episodic LH secretion are associated with reduced rates of ovulation in humans and nonhuman primates.58, 59, 60
LATE FOLLICULAR PHASE
In the late follicular phase, the diameter of the selected follicle increases exponentially to a final size of 15–20 mm, and as a result secretion of estradiol increases exponentially. Vascular development in the dominant follicle plays a crucial role in this process because short-term inhibition of angiogenesis after anti-VEGF antibody administration during the later growth phase of the dominant follicle interferes with its normal development, interrupts the characteristic rise in estradiol secretion, and results in a significant lengthening of the follicular phase.61 Under thess conditions, access to peripheral factors needed to support follicle growth, such as the gonadotropins, is most probably limited.
The increased estrogen milieu in the late follicular phase modifies the genital tract.62 The glandular endometrium proliferates, the characteristics of cervical mucus change (increased secretion, decreased viscosity, and increased pH), and cornification of the vaginal epithelium occurs.
As the dominant follicle approaches maturity, estradiol secretion reaches its peak. This peak acts as the crucial ovarian signal that triggers the ovulatory gonadotropin surge. Experimental neutralization of this estradiol signal results in a suppression of the gonadotropin surge.63 In humans, an LH surge can be induced experimentally during the early follicular phase after the administration of estradiol in amounts mimicking those seen in the late follicular phase (see Fig. 5B). The process by which estrogen transients stimulate the release of LH and FSH is referred to as the positive estradiol feedback loop and represents the crucial process that synchronizes follicle maturity and ovulation.
Whether gonadotropins are released after an estradiol challenge depends on the strength and duration of the estrogen signal. Minimal requirements are an increase in circulating estradiol concentrations to a threshold level close to that seen spontaneously in the late follicular phase and for a period of at least 34 hours. Subthreshold (even maintained for a prolonged period) or short-lived (even larger than threshold) rises in estradiol levels do not elicit an LH surge experimentally.64, 65
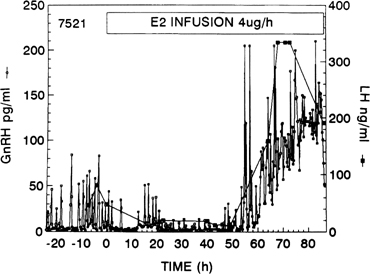
As shown in nonhuman primates and in sheep, the preovulatory gonadotropin surge is preceded by a dramatic and sustained rise in GnRH (Fig. 10).66, 67, 68 Increased GnRH secretion drives the preovulatory LH surge in a dose-dependent fashion. These data suggest a substantial central action of estradiol on the hypothalamus, possibly on different neuronal cell populations than those modulating the estradiol negative feedback loop.69 Experimental evidence indicates that the preovulatory LH surge depends on GnRH stimulation throughout its entire course, but that the GnRH surge persists many hours beyond the termination of the LH surge (Fig. 11).70 The LH surge terminates even though there seems to be no change in the biologic activity of GnRH.71 The functional significance of this excess of GnRH to midcycle events remains to be determined. Suggesting a similar role of GnRH in the induction of the preovulatory gonadotropin surge in humans is the observation that administration of a GnRH antagonist Nal-Glu acutely inhibits the LH surge and ovulation.72 Because LH responses to identical GnRH stimuli increase during the follicular phase in parallel with increasing estradiol concentrations, estradiol also may act to augment the gonadotrope’s responsiveness to GnRH.73 It is logical to postulate that during the normal menstrual cycle, central and pituitary sites respond to the estradiol positive feedback loop signal to ensure a timely gonadotropin surge.
Although progesterone secretion is minimal during the follicular phase, there is a small but significant rise in this hormone at the time of initiation of the gonadotropin surge (see below). In low concentrations, progesterone facilitates LH release and it is believed that this increase in progesterone may be required for the full expression of the gonadotropin surge because administration of a progesterone antagonist at midcycle in humans results in a delay or in the abolition of the gonadotropin surge.74, 75
OVULATORY PERIOD
The estradiol-induced gonadotropin surge initiates a chain of events that includes a series of finely orchestrated biochemical events, many of which are still poorly understood, and that culminates in follicular rupture and ovulation. Hormonally, drastic changes in ovarian steroid profiles occur after alterations of enzymatic activity. The large increase in LH inhibits androgen production, and as a result estradiol concentrations decrease drastically from the preovulatory peak. Granulosa cells become 'luteinized', and consequently a small preovulatory rise in progesterone occurs within one hour of the LH surge (see Fig. 1).
Mechanically, ovulation consists of a rapid follicular enlargement with subsequent protrusion of the follicle from the ovarian surface. About 24–36 hours after the initiation of the gonadotropin surge or 18 hours after the gonadotropin peak, follicle rupture results in the expulsion of an oocyte-cumulus complex. Rupture does not seem to be caused by an increase in intrafollicular pressure; rather the LH surge induces an increase in follicular volume, which is related to an increase in follicular blood flow, a decrease in vascular permeability, and subsequent changes in the properties of the follicular wall.76 These changes in vascular function most probably reflect the large local concentrations of angiogenic factors in the mature follicle and a functional VEGF system.77 The LH surge stimulates a cascade of ovarian gene expression in the preovulatory follicle78 resulting in an acute inflammatory-like process which generates an increase in proteolytic enzymes, such as plasminogen activator, plasmin, and matrix metalloproteinases, which bring about the degradation of the perifollicular matrix.79 Pharmacologic blockage of any of these enzymes results in a reduction in the ovulation rate. The periovulatory follicle produces prostaglandins in response to the LH surge, and these also seem to be crucial to follicle rupture, although their direct role in primates remains to be shown.80 A paracrine role of progesterone in the mediation of LH effects on follicular rupture has been suggested in lower species.81
The luteal phase
Shortly after the ovulatory gonadotropin surge, the granulosa cells of the ovulated dominant follicle fold, the basal lamina, which separates the granulosa and theca layers, is disrupted, and capillaries from the theca interna invade the granulosa layer (which until now had been avascular) to form such an extensive capillary network that each steroidogenic cell is now close to microvascular elements. Thus, after ovulation, a new ovarian structure emerges, the corpus luteum. New key steroidogenic enzymes are activated so that the hallmark of the human corpus luteum is its secretion of both progesterone and estradiol (see Fig. 1). Significantly, the passage of granulosa and theca cells of the now defunct dominant follicle into progesterone secreting cells is a terminal cell differentiation in that it is associated with an arrest in cell proliferation. The corpus luteum also secretes significant amounts of inhibin A (see Fig. 8).
The mammalian corpus luteum is an exceptionally dynamic organ in which growth and development occur rapidly. As for the follicle, vascular growth plays a central role in this process. Angiogenic factors, such as VEGF, are present in high quantity in the forming and developing corpus luteum.82, 83 In monkeys, experimental treatments that interfere with normal VEGF activity in the early and mid luteal phase suppress the intense luteal endothelial proliferation and vascular development; as a result, luteal function is compromised, as indicated by a marked fall in plasma progesterone levels.84, 85 It is well documented that corpus luteum function depends primarily on pituitary LH secretion throughout the luteal phase.86, 87, 88 Studies in hypophysectomized women, in whom ovulation was induced by LH treatment, indicate that continuous administration of small amounts of LH is essential to maintain the viability of the corpus luteum.30 GnRH antagonist treatment readily disrupts luteal cell morphology and suppresses plasma progesterone.89 The above experimental results indicate that LH and its support of local VEGF are as crucial to corpus luteum function as they are to follicular function.
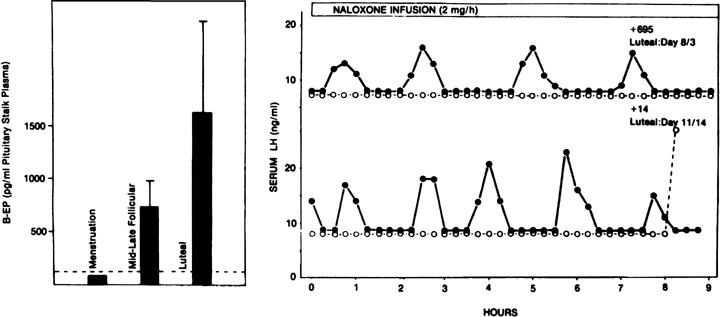
Progesterone dominance in the luteal phase results in a significant decrease in GnRH/LH pulse frequency throughout this stage of the cycle (see Fig. 7).90 There is good experimental evidence to indicate that this is an effect by progesterone which is mediated by central opioid peptides, because administration of competitive antagonists to endogenous opiates, such as naloxone, is particularly effective in increasing LH pulse frequency (Fig. 12B).91, 92, 93, 94, 95 A relevant endogenous opiate known to inhibit pulsatile LH secretion is β-endorphin. Its neuronal cell bodies are preferentially concentrated in the arcuate nucleus, an area known to be involved in the control of gonadotropin secretion. β-Endorphin release from the hypothalamus reflects the ovarian endocrine milieu. In the absence of significant concentrations of ovarian steroids, such as after ovariectomy or at menstruation, β-endorphin release is lowest, whereas it is highest in the presence of estradiol and progesterone, such as during the luteal phase (Fig. 12A).96, 97 The naloxone test is an indirect indicator of central opiate activity.
|
Progesterone also affects the hypothalamic thermoregulatory center so that an increase in basal body temperature (BBT) accompanies increased progesterone secretion during the luteal phase, giving rise to the typical BBT curve of the ovulatory menstrual cycle. Progesterone dominance during this phase of the menstrual cycle also modifies the genital tract in preparation for possible implantation of a fertilized ovum; there is increased secretory activity by the endometrial glands and changes in the characteristics of the cervical mucus, which is now thick and viscous. At its large luteal concentrations, progesterone also inhibits the estradiol positive feedback loop98 and gametogenic follicle growth.99
In primates, the life span of the corpus luteum is limited to a period of about 14 days. According to biochemical and histologic criteria, the corpus luteum reaches maturity 8–9 days after ovulation, after which time its secretory capability declines. Midway through the luteal phase, levels of estradiol and progesterone decrease, and menstruation follows ovulation by 13–15 days. Structural luteolysis is a complex process responsible for the elimination of the corpus luteum. Steroidogenic luteal cells undergo characteristic degenerative changes, with intense cytoplasmic vacuolization and invasion by macrophages followed by a perimenstrual apoptotic wave.100 The factors responsible for luteolysis in primates remain to be identified.101 It is now believed that regression of the corpus luteum may be related to an alteration in age-dependent luteal cell responsiveness and is dictated by various luteotropic and luteolytic agents, the existence and dynamics of which have not been investigated. Only rapidly rising concentrations of chorionic gonadotropin (hCG) can rescue the corpus luteum. Although uterine prostaglandin F2α seems to be an important luteolytic signal in nonprimate species, the primate uterus is not the source of luteolytic agents because hysterectomy does not result in a prolonged luteal phase.101
After the dramatic decrease in estradiol and progesterone secretion at the end of the luteal phase, there is a characteristic divergence in the ratio of the two gonadotropins, now favoring a specific rise in FSH (see Fig. 2). The precise reason for the increase in the FSH:LH ratio at the end of the cycle remains to be determined, but there are several possibilities, all of which may play a role to a certain degree. Because FSH seems to be slightly more sensitive to the estradiol negative feedback loop than LH,102 a rise in FSH may be the result of the rapid decline in estradiol at that time. The rise in FSH also may reflect differential effects of GnRH pulse frequency on the synthesis of LH and FSH, as a lower pulse frequency favors FSH β subunit synthesis over that of the LH subunit, and a larger pool of FSH would be available for release at the end of this period (see above). It also has been speculated that the accompanying fall in inhibin A (from a peak in the mid luteal phase)103 may play a role in initiating the intercycle FSH rise. The increase in the FSH:LH ratio heralds the new cycle and the recruitment of a new cohort of follicles.
PATHOPHYSIOLOGY OF THE MENSTRUAL CYCLE
As outlined in the preceding sections, the normal ovulatory menstrual cycle requires a remarkable coordination between all components of the hypothalamic-pituitary-gonadal axis and other target tissues. The ovarian steroids, through their positive and negative feedback loops, play a crucial role by regulating gonadotropin levels. Cyclic dysfunction may be related to abnormalities in estradiol function or may reflect external influences, which, for example, may interfere with the normal function of the GnRH pulse generator. It may include a range of changes from 'qualitative' changes in the cycle, such as in the inadequate luteal phase syndrome, to a cessation of cyclic activity, such as in amenorrhea.
Abnormalities of the hypothalamic-pituitary unit
A properly active GnRH pulse generator is essential for normal gonadotropin release and for a normal ovulatory menstrual cycle to occur. Conditions that prevent or interfere with the function of the pulse generator disrupt the pituitary-ovarian axis and the cycle. Absence of GnRH release, such as observed in Kallmann’s syndrome, in which the migration of the GnRH neuron is not completed and contact with the hypophyseal portal veins is not established (see earlier),8 leads to idiopathic hypogonadotropic hypogonadism and delayed puberty. This primary hypothalamic amenorrhea syndrome also is characterized by anosmia, probably reflecting the initial defect in paraolfactory areas.
Frequent causes of cyclic dysfunction are related to lifestyle variables, such as psychogenic stress, and exercise-related or diet-related causes that affect hypothalamic function. The resultant condition frequently is referred to as functional hypothalamic amenorrhea or functional hypothalamic chronic anovulation.104 The syndrome is characterized by a significant reduction in the activity of the GnRH pulse generator resulting in a decrease in the frequency of pulsatile LH release. Because adherence to a specific regimen of GnRH and LH pulse frequency is crucial for normal menstrual cyclicity and reproductive function (see earlier), such abnormal episodic LH secretion can lead to anovulation.58 The degree of inhibition of the GnRH pulse generator determines whether the menstrual cycle is disturbed only slightly or whether the subject is anovulatory. One remarkable aspect of this syndrome is, however, that in most cases, it is readily reversible upon removal of the cause.
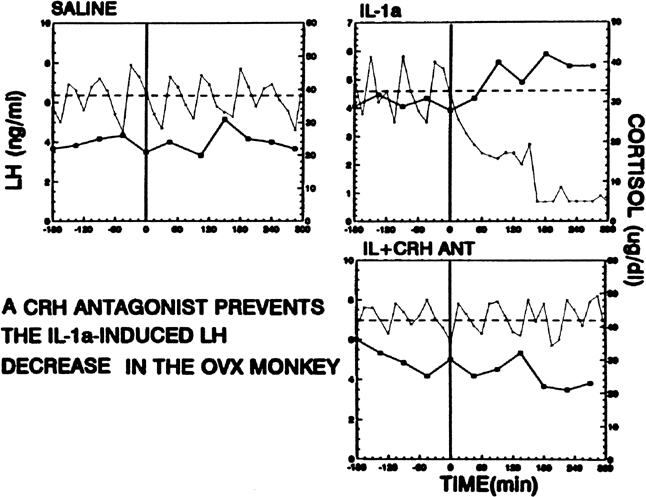
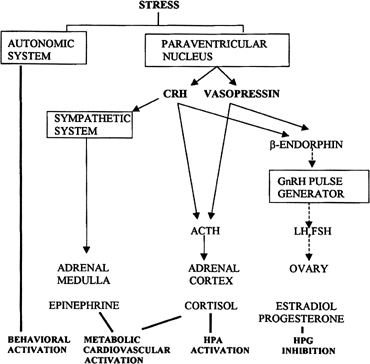
Psychological or environmental stress has long been reported to produce chronic amenorrhea.104, 105, 106 The observation that in this situation cortisol secretion is higher than normal suggests an association between increased hypothalamic-pituitary-adrenal (HPA) axis activity and reduced GnRH drive.104 Experimental data in nonhuman primates support the concept that stress-induced functional hypothalamic amenorrhea develops in response to alterations in hypothalamic function.105, 106 A primary role for corticotropin-releasing hormone (CRH), the principal neurohormone of the HPA axis, in inhibiting the GnRH pulse generator is now well known. For instance, the acute inhibitory effects on pulsatile LH secretion of an immune stress challenge known to activate the HPA axis can be prevented by the administration of a CRH antagonist (Fig. 13).107 Vasopressin, a peptide frequently co-packaged with CRH within CRH-containing secretory granules and co-released with CRH in stress, especially in chronic stress,108, 109 also can inhibit pulsatile LH secretion, and administration of a vasopressin antagonist also reverts the acute inhibitory effects of an immune stress challenge on LH.110 Although CRH or vasopressin administration increases adrenocorticotropic hormone and cortisol secretion, the ensuing acute gonadotropin inhibition is unrelated to these hormones in the primate. LH inhibition after CRH is observed in adrenalectomized monkeys and cannot be mimicked by the infusion of adrenocorticotropic hormone (ACTH) or cortisol, suggesting a central site of action of the HPA neurohormones.111, 112 This action is modulated by central endogenous opiates.113, 114 A simplified scheme of the central response to stress and the inhibition of the HPA axis is illustrated in Fig. 14.
|
|
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Energetics and reproduction are linked115 and dietary restriction may suppress pulsatile LH release and affect the menstrual cycle, suggesting that the GnRH pulse generator is modulated by metabolic fuels and energy-related peptides.116, 117, 118, 119, 120, 121 The exact metabolic clues that signal the GnRH pulse generator of subtle changes in the metabolic status are presently being investigated. Studies in monkeys indicate that the clue depends on caloric intake but not on changes in body mass or composition and not on intake of a specific nutrient or in plasma glucose or insulin concentrations.122 A role for energy-related peptides in these processes is certainly called for. For example, data on the adipose tissue-derived hormone leptin123 indicate that leptin levels decrease with fasting or undernutrition in parallel with decreased gonadotropins.124 Observations in animals suggest that leptin may function as a metabolic signal to the neuroendocrine reproductive system and that under conditions of inadequate energy reserves, low leptin levels may act as a metabolic 'gate' to inhibit the neuroendocrine reproductive axis.125 In support of this statement is the observation that leptin treatment in fasted animals prevents the decrease in LH pulse frequency that is seen after fasting, indicating that this hormone conveys information about nutrition to mechanisms controlling neuroendocrine function.126 Ghrelin levels, another energy-related peptide derived from the stomach,127 have been shown to increase during negative energy balance128 while an acute ghrelin infusion significantly decreases LH pulse frequency in the monkey.129
The extreme prototype of nutrition-related functional hypothalamic amenorrhea is the anorexia nervosa syndrome, which presents as a classic triad of amenorrhea, weight loss (sometimes to emaciation), and behavioral changes. The syndrome is characterized by a hypogonadotropic state (although FSH may be less affected than LH) and, in extreme cases, a regression to a prepubertal pattern of LH secretion, with hypoestrogenism.104, 116 With recovery, LH pulsatility returns, first mimicking the pubertal pattern characterized by a nocturnal increase in pulsatility, then an adult pattern. High ghrelin levels are reported in reported in this syndrome.130 Other syndromes associated with weight loss and altered reproductive function include bulimia, weight loss of 10–15% below ideal body weight, and various diets.116
GnRH pulse activity also may be affected by strenuous exercise, including jogging and athletics.131, 132 Depending on the intensity of the exercise, cyclic abnormalities may progress to a hypoestrogenic hypogonadotropic pattern. Physical activity alone may not be sufficient to cause amenorrhea, however, and other adjunct factors, such as low body weight, change in diet, weight loss, or the perceived stress of the exercise, may synergize to cause the effect.
Abnormalities of the hormonal feedback loops
Abnormal feedback conditions may result from an inappropriate extraglandular source of estrogen. In women, most of the circulating estrogens derive from estradiol secreted by the ovarian follicle or corpus luteum. Some estrogens also derive from peripheral conversion of androstenedione to estrone in adipose tissue and skin. In several pathologic conditions, such as congenital adrenal hyperplasia, Cushing’s syndrome, and androgen-producing tumors of either adrenal or ovarian origin, estrogen precursor availability is increased, and extraglandular production of estrogen may become excessive.133 The androgen-to-estrogen conversion rate also increases with advancing age and in obese patients. Because this estrogen increase is acyclic and not under the control of gonadotropins, the original estradiol feedback signal may be distorted or masked, and cyclic irregularities may occur. In the polycystic ovary syndrome, which combines metabolic and hormonal disturbances, androgen production also is enhanced; LH pulse frequency, or in some cases LH pulse amplitude, is increased in greater than 60% of the patients.134, 135 Accelerated LH pulse frequency in women with polycystic ovary syndrome is not influenced by body mass index136 and most probably represents a basic component of hypothalamic dysfunction.137, 138 Since an increased GnRH pulse frequency favors LH synthesis (see above), there is a resultant increased LH:FSH ratio. Abnormal steroid production and further disturbances in feedback regulation may occur.
In a few patients, abnormally low gonadotropin secretion has been attributed to an increase in the sensitivity of the hypothalamic GnRH pulse generator to the estradiol negative feedback loop.139 Shifts in feedback sensitivity to estradiol have been suggested to form the basis for seasonality in the reproductive process in sheep: seasonal photic information is relayed to the pineal gland, which transduces the message into a hormonal signal in the form of melatonin. The circadian rhythm of melatonin, variable with the season, determines the capacity of the GnRH pulse generator to respond to the negative feedback action of estradiol, and LH pulse patterns inductive or suppressive of normal cyclicity are set.140
During the normal menstrual cycle, the estradiol positive feedback loop ensures the coupling of follicular maturity with the pituitary gonadotropin surge (the stimulus to ovulation). Delayed estradiol increments or insensitivity of the hypothalamic-pituitary axis to the positive estrogen feedback loop (as may occur during the first pubertal cycles) may delay the LH surge beyond the time that ovulation is possible. Premature estradiol increments or increased hypothalamic-pituitary sensitivity to the estrogen stimulus may induce a premature LH surge, which may interfere with or arrest follicular maturation. Experimental evidence in monkeys and humans shows that certain stress stimuli, when occurring in the presence of midfollicular phase levels of estradiol, can stimulate rather than inhibit LH release.106, 141, 142 Thus, it may be that a 'stress' episode occurring during the follicular phase could affect the menstrual cycle by inducing a gonadotropin surge at an inappropriate time of the cycle, interfering with proper ovarian-pituitary signals.
Abnormalities of the pituitary
Defects in menstrual cyclicity may be caused by pituitary lesions, which may result in trophic hormone deficiency, or by pituitary tumors. A typical pituitary lesion is the one that occurs in conjunction with peripartum or postpartum hemorrhage and ischemic shock (Sheehan’s syndrome). Depending on the extent of trophic hormone deficiency, symptoms of gonadotropin, thyroid, or adrenal hormone secretory defects may coexist. The most common type of pituitary tumor disrupting the menstrual cycle is the prolactin-secreting adenoma causing the amenorrhea-galactorrhea syndrome.143 Through central mechanisms that are not yet known, hyperprolactinemia suppresses the GnRH pulse generator and pulsatile LH and FSH release. Hypothalamic control of prolactin is predominantly inhibitory, and the hypothalamic factor responsible for controlling this hormone is the neurotransmitter dopamine. In this case, dopamine is released from high-density dopamine nerve endings in the median eminence into the hypothalamic-hypophyseal vessels. The functional aberrations in prolactin and in pulsatile LH and FSH secretion in this syndrome can be reversed readily by the administration of dopamine agonists.
Abnormalities of the corpus luteum
Another example of a frequent cyclic abnormality is the inadequate luteal phase syndrome, which is characterized by blunted progesterone secretion throughout the luteal phase.144 In turn, the lack of progesterone interferes with the proper luteal endometrial sequence. The resultant abnormal secretory activity of the endometrium and other uterine deficiencies related to insufficient progesterone levels may prevent the normal implantation process and may result in infertility. Because the corpus luteum derives entirely from the follicle that has matured during the follicular phase of that cycle, abnormal events that occur in the early follicular phase may influence luteal function. Inappropriate patterns of FSH secretion early in the follicular phase may lead to deficiencies in the development of the graafian follicle and in estradiol secretion, which may result in the inappropriate luteinization of the granulosa cells and a smaller sized corpus luteum with less secretory capability.145, 146
Relevant observations in experimental stress models in nonhuman primates suggest that the first clinical manifestation of cyclic dysfunction in an otherwise normal ovulatory primate subjected to stress usually occurs in the form of a significant reduction in integrated progesterone concentrations during the luteal phase, for example in non-human primates subjected to short inflammatory-like or psychogenic stress challenges.147, 148, 149 A similar prevalence of luteal defect in cycling women has been reported as a result of stress, exercise, or eating disorders.150, 151, 152, 153
REFERENCES
Ferin M, Jewelewicz R, Warren M: The menstrual cycle: Physiology, reproductive disorders and infertility. New York. Oxford Press, 1993 |
|
Ferin M: The menstrual cycle: an integrative view. In: Reproductive Endocrinology, Surgery, and Technology. Adashi EY, Rock JA, Rosenwaks Z (eds). Raven Press NY, chapter 6: 103, 1996 |
|
Rangaraju NS, Xu JF, Harris RB: Pro-gonadotropin-releasing hormone protein is processed within hypothalamic neurosecretory granules. Neuroendocrinology. 1991 Jan;53(1):20-8. |
|
Herbison AE: Physiology of the gonadotropin-releasing hormone neuronal network. In: Physiology of reproduction. Neill JD (ed), Elsevier-Academic Press, chapter 28:1415,2006 |
|
Hotchkiss J, Knobil E: The hypothalamic pulse generator: The reproductive core. Adashi EY, Rock JA, Rosenwaks Z (eds): Reproductive Endocrinology, Surgery, and Technology. Lipincott, chapter 7:123, 1996. |
|
Schwanzel-Fukuda M, Morrell JI, Pfaff DW: Ontogenesis of neurons producing luteinizing hormone-releasing hormone (LHRH) in the nervus terminalis of the rat. J Comp Neurol. 1985 Aug 15;238(3):348-64. |
|
Seminara SB, Hayes FJ, Crowley WF Jr: Gonadotropin-releasing hormone deficiency in the human (idiopathic hypogonadotropic hypogonadism and Kallmann's syndrome): pathophysiological and genetic considerations. Endocr Rev. 1998 Oct;19(5):521-39. |
|
Schwanzel-Fukuda M, Bick D, Pfaff DW: Luteinizing hormone-releasing hormone (LHRH)-expressing cells do not migrate normally in an inherited hypogonadal (Kallmann) syndrome. Brain Res Mol Brain Res. 1989 Dec;6(4):311-26. |
|
Tobet SA, Schwarting GA: Minireview: recent progress in gonadotropin-releasing hormone neuronal migration. Endocrinology. 2006 Mar;147(3):1159-65. Epub 2005 Dec 22. |
|
Lee JM, Tiong J, Maddox DM et al: Temporal migration of gonadotrophin-releasing hormone-1 neurones is modified in GAD67 knockout mice. J Neuroendocrinol. 2008 Jan;20(1):93-103. |
|
Cariboni A, Hickok J, Rakic S et al: Neuropilins and their ligands are important in the migration of gonadotropin-releasing hormone neurons. J Neurosci. 2007 Feb 28;27(9):2387-95. |
|
Nielsen-Preiss SM, Allen MP, Xu M et al: Adhesion-related kinase induction of migration requires phosphatidylinositol-3-kinase and ras stimulation of rac activity in immortalized GnRH neuronal cells. Endocrinology. 2007 Jun;148(6):2806-14. Epub 2007 Mar 1. |
|
Carmel PW, Araki S, Ferin M: Pituitary stalk portal blood collection in rhesus monkeys: evidence for pulsatile release of GnRH. Endocrinology. 1976 Jul;99(1):243-8. |
|
Moenter SM, Brand RC, Karsch FJ: Dynamics of gonadotropin-releasing hormone (GnRH) secretion during the GnRH surge: insights into the mechanism of GnRH surge induction. Endocrinology. 1992 May;130(5):2978-84. |
|
Terasawa E, Keen KL, Mogi K et al: Pulsatile release of luteinizing hormone-releasing hormone (LHRH) in cultured LHRH neurons derived from the embryonic olfactory placode of the rhesus monkey. Endocrinology. 1999 Mar;140(3):1432-41. |
|
Dungan HM, Clifton DK, Steiner RA: Minireview: kisspeptin neurons as central processors in the regulation of GnRH secretion. Endocrinology. 2006 Mar;147(3):1154-8. Epub 2005 Dec 22. |
|
Plant TM, Witchel SF: Puberty in nonhuman primates and humans. In: Physiology of reproduction. Neill JD (ed), Elsevier-Academic Press, chapter 40: 2177, 2006 |
|
Ross JL, Loriaux DL, Cutler GB Jr: Developmental changes in neuroendocrine regulation of gonadotropin secretion in gonadal dysgenesis. J Clin Endocrinol Metab. 1983 Aug;57(2):288-93. |
|
Soules MR, Steiner RA, Cohen NL et al: Nocturnal slowing of pulsatile luteinizing hormone secretion in women during the follicular phase of the menstrual cycle. J Clin Endocrinol Metab. 1985 Jul;61(1):43-9. |
|
Bousfield JR, Jia l, Ward DN: Gonadotropins: chemistry and biosynthesis. In: Physiology of reproduction. Neill JD (ed), Elsevier-Academic Press, 2006, chapter 30:1581-1634 |
|
Southworth MB, Matsumoto AM, Gross KM et al: The importance of signal pattern in the transmission of endocrine information:pituitary gonadotropin responses to continuous and pulsatile GnRH. J Clin Endocrinol Metab. 1991 Jun;72(6):1286-9. |
|
Belchetz PE, Plant TM, Nakai Y et al: Hypophysial responses to continuous and intermittent delivery of hypopthalamic GnRH. Science. 1978 Nov 10;202(4368):631-3. |
|
Barbieri RL: Clinical applications of GnRH and its analogues. Trends Endocrinol Metab. 1992 Jan-Feb;3(1):30-4. |
|
Jeong KH, Kaiser UB: Gonadotropin-releasing hormone regulation of gonadotropin biosynthesis and secretion. In: Physiology of reproduction. Neill JD (ed), Elsevier-Academic Press, chapter 31: 1635,2006. |
|
Haisenleder DJ, Dalkin AC, Ortolano GA et al: A pulsatile gonadotropin-releasing hormone stimulus is required to increase transcription of the gonadotropin subunit genes: evidence for differential regulation of transcription by pulse frequency in vivo. Endocrinology. 1991 Jan;128(1):509-17. |
|
Spratt DI, Finkelstein JS, Butler JP et al: Effects of increasing the frequency of low doses of gonadotropin-releasing hormone on gonadotropin secretion in GnRH-deficient men. J Clin Endocrinol Metab. 1987 Jun;64(6):1179-86. |
|
Wildt L, Hausler A, Marshall G et al: Frequency and amplitude of gonadotropin-releasing hormone stimulation and gonadotropin secretion in the rhesus monkey. Endocrinology. 1981 Aug;109(2):376-85. |
|
Gross KM, Matsumoto AM, Bremner WJ: Differential control of luteinizing hormone and follicle-stimulating hormone secretion by luteinizing hormone-releasing hormone pulse frequency in man. J Clin Endocrinol Metab. 1987 Apr;64(4):675-80. |
|
Yamaji T, Dierschke DJ, Bhattacharya AN et al: The negative feedback control by estradiol and progesterone of LH secretion in the ovariectomized rhesus monkey. Endocrinology. 1972 Mar;90(3):771-7. |
|
Vande Wiele RL, Bogumil J, Dyrenfurth I et al: Mechanisms regulating the menstrual cycle in women. Recent Prog Horm Res. 1970;26:63-103. |
|
Yen SS, Tsai CC, Vandenberg G et al: Gonadotropin dynamics in patients with gonadal dysgenesis: a model for the study of gonadotropin regulation. J Clin Endocrinol Metab. 1972 Dec;35(6):897-904. |
|
Nippoldt TB, Reame NE, Kelch RP et al: The roles of estradiol and progesterone in decreasing luteinizing hormone pulse frequency in the luteal phase of the menstrual cycle. J Clin Endocrinol Metab. 1989 Jul;69(1):67-76. |
|
Burger HG, Igarashi M: Inhibin: definition and nomenclature, including related substances. J Clin Endocrinol Metab. 1988 Apr;66(4):885-6. |
|
Groome NP, Illingworth PJ, O'Brien M et al: Measurement of dimeric inhibin B throughout the human menstrual cycle. J Clin Endocrinol Metab. 1996 Apr;81(4):1401-5. |
|
Ying SY: Inhibins, activins, and follistatins: gonadal proteins modulating the secretion of FSH. Endocr Rev. 1988 May;9(2):267-93. |
|
Fraser HM, Groome NP, McNeilly AS: Follicle-stimulating hormone-inhibin B interactions during the follicular phase antagonist and antiestrogen treatment. J Clin Endocrinol Metab. 1999 Apr;84(4):1365-9. |
|
Welt CK, McNicholl DJ, Taylor AE et al: Female reproductive aging is marked by decreased secretion of dimeric inhibin. J Clin Endocrinol Metab. 1999 Jan;84(1):105-11. |
|
Reame NE, Wyman TL, Phillips DJ et al: Net increase in stimulatory input resulting from a decrease in inhibin B and an increase in activin A may contribute in part to the rise in follicular phase FSH of aging cycling women. J Clin Endocrinol Metab. 1998 Sep;83(9):3302-7. |
|
Welt CK, Hall JE, Adams JM et al: Relationship of estradiol and inhibin to the follicle-stimulating hormone variability in hypergonadotropic hypogonadism or premature ovarian failure. J Clin Endocrinol Metab. 2005 Feb;90(2):826-30. Epub 2004 Nov 23. |
|
McGee EA, Hsueh AJ: Initial and cyclic recruitment of ovarian follicles. Endocr Rev. 2000 Apr;21(2):200-14. |
|
Goodman AL, Hodgen GD: The ovarian triad of the primate menstrual cycle. Recent Prog Horm Res. 1983;39:1-73. |
|
Zeleznik AJ: Follicle selection in primates: "many are called but few are chosen". Biol Reprod. 2001 Sep;65(3):655-9. |
|
Burger HG, Groome NP, Robertson DM: Both inhibin A and B respond to exogenous follicle-stimulating hormone in the follicular phase of the human menstrual cycle. J Clin Endocrinol Metab. 1998 Nov;83(11):4167-9. |
|
Eldar-Geva T, Robertson DM, Cahir N et al: Relationship between serum inhibin A and B and ovarian follicle development after a daily fixed dose administration of recombinant FSH.J Clin Endocrinol Metab. 2000 Feb;85(2):607-13. |
|
Hsueh AJ, Eisenhauer K, Chun SY et al: Gonadal cell apoptosis. Recent Prog Horm Res. 1996;51:433-55; discussion 455-6. |
|
Goodman AL, Nixon WE, Johnson DK et al: Regulation of folliculogenesis in the cycling rhesus monkey: selection of the dominant follicle. Endocrinology. 1977 Jan;100(1):155-61. |
|
Zeleznik AJ, Schuler HM, Reichert LE Jr: Gonadotropin-binding sites in the rhesus monkey ovary: role of the vasculature inthe selective distribution of human chorionic gonadotropin to the preovulatory follicle. Endocrinology. 1981 Aug;109(2):356-62. |
|
Yamamoto S, Konishi I, Tsuruta Y et al: Expression of vascular endothelial growth factor (VEGF) during folliculogenesis and corpus luteum formation in the human ovary. Gynecol Endocrinol 1997 Dec;11 (6):371-81 |
|
Carmeliet P: Mechanisms of angiogenesis and arteriogenesis. Nat Med. 2000 Apr;6(4):389-95. |
|
Zimmermann RC, Xiao E, Bohlen P et al: Administration of antivascular endothelial growth factor receptor 2 antibody in the early follicular phase delays follicular selection and development in the rhesus monkey. Endocrinology. 2002 Jul;143(7):2496-502. |
|
Hazzard TM, Molskness TA, Chaffin CL et al: Vascular endothelial growth factor (VEGF) and angiopoietin regulation by gonadotrophin and steroids in macaque granulosa cells during the peri-ovulatory interval. Mol Hum Reprod. 1999 Dec;5(12):1115-21. |
|
Sullivan MW, Stewart-Akers A, Krasnow JS et al: Ovarian responses in women to recombinant follicle-stimulating hormone andluteinizing hormone (LH): a role for LH in the final stages of follicularmaturation. J Clin Endocrinol Metab. 1999 Jan;84(1):228-32. |
|
Simpson ER: Estrogens. In: Adashi EY, Rock JA, Rosenwaks Z (eds): Reproductive Endocrinology, Surgery, and Technology. Philadelphia, Lipincott-Raven, chapter 23:477,1996. |
|
Zeleznik AJ, Hutchison JS, Schuler HM: Interference with the gonadotropin-suppressing actions of estradiol in macaques overrides the selection of a single preovulatory follicle. Endocrinology. 1985 Sep;117(3):991-9. |
|
Ferin M, Dyrenfurth I, Cowchock S et al: Active immunization to 17 beta-estradiol and its effects upon the reproductive cycle of the rhesus monkey. Endocrinology. 1974 Mar;94(3):765-76. |
|
Giudice LC: Insulin-like growth factors and ovarian follicular development. Endocr Rev. 1992 Nov;13(4):641-69. |
|
Webb R, Nicholas B, Gong JG et al: Mechanisms regulating follicular development and selection of the dominant follicle. Reprod Suppl. 2003;61:71-90. |
|
Filicori M, Tabarelli C, Casadio P et al: Interaction between menstrual cyclicity and gonadotropin pulsatility. Horm Res. 1998;49(3-4):169-72. |
|
Filicori M, Cognigni G, Dellai P et al: Role of gonadotrophin releasing hormone secretory dynamics in the control of the human menstrual cycle. Hum Reprod. 1993 Nov;8 Suppl 2:62-5. |
|
Knobil E, Plant TM, Wildt L et al: Control of the rhesus monkey menstrual cycle: permissive role of hypothalamic GnRH. Science. 1980 Mar 21;207(4437):1371-3. |
|
Zimmermann RC, Xiao E, Husami N et al: Short-term administration of antivascular endothelial growth factor antibody in the late follicular phase delays follicular development in the rhesus monkey. J Clin Endocrinol Metab. 2001 Feb;86(2):768-72. |
|
Giudice LC: The endometrial cycle. In: Adashi EY, Rock JA, Rosenwaks Z (eds): Reproductive Endocrinology, Surgery, and Technology. Philadelphia, Lipincott-Raven, chapter 13: 272, 1996. |
|
Schwartz U, Dyrenfurth I, Khalaf S et al: A comparison of the effects of active immunization of female rhesus monkeys to estradiol-17 or progesterone-20 conjugates. J Steroid Biochem. 1975 Mar-Apr;6(3-4):541-5. |
|
Fritz MA, McLachlan RI, Cohen NL et al: Onset and characteristics of the midcycle surge in bioactive and immunoactive luteinizing hormone secretion in normal women: influence of physiologicalvariations in periovulatory ovarian steroid hormone secretion. J Clin Endocrinol Metab. 1992 Aug;75(2):489-93. |
|
Karsch FJ, Weick RF, Butler WR et al: Induced LH surges in the rhesus monkey: strength-duration characteristics of the estrogen stimulus. Endocrinology. 1973 Jun;92(6):1740-7. |
|
Moenter SM, Caraty A, Locatelli A et al: Pattern of gonadotropin-releasing hormone (GnRH) secretion leading up to ovulation in the ewe: existence of a preovulatory GnRH surge. Endocrinology. 1991 Sep;129(3):1175-82. |
|
Xia L, Van Vugt D, Alston EJ et al: A surge of gonadotropin-releasing hormone accompanies the estradiol-induced gonadotropin surge in the rhesus monkey. Endocrinology. 1992 Dec;131(6):2812-20. |
|
Pau KY, Berria M, Hess DL et al: Preovulatory gonadotropin-releasing hormone surge in ovarian-intact rhesus macaques. Endocrinology. 1993 Oct;133(4):1650-6. |
|
Caraty A, Fabre-Nys C, Delaleu B et al: Evidence that the mediobasal hypothalamus is the primary site of action ofestradiol in inducing the preovulatory gonadotropin releasing hormone surge in the ewe. Endocrinology. 1998 Apr;139(4):1752-60. |
|
Evans NP, Dahl GE, Caraty A et al: How much of the gonadotropin-releasing hormone (GnRH) surge is required for generation of the luteinizing hormone surge in the ewe? Duration of the endogenous GnRH signal. Endocrinology. 1996 Nov;137(11):4730-7. |
|
Caraty A, Antoine C, Delaleu B et al: Nature and bioactivity of gonadotropin-releasing hormone (GnRH) secreted during the GnRH surge. Endocrinology. 1995 Aug;136(8):3452-60. |
|
Ditkoff EC, Cassidenti DL, Paulson RJ et al: The gonadotropin-releasing hormone antagonist (Nal-Glu) acutely blocks the luteinizing hormone surge but allows for resumption of folliculogenesis in normal women. Am J Obstet Gynecol. 1991 Dec;165(6 Pt 1):1811-7. |
|
Lasley BL, Wang CF, Yen SS: The effects of estrogen and progesterone on the functional capacity of the gonadotrophs. J Clin Endocrinol Metab. 1975 Nov;41(5):820-6. |
|
Liu JH, Yen SS: Induction of midcycle gonadotropin surge by ovarian steroids in women: a critical evaluation. J Clin Endocrinol Metab. 1983 Oct;57(4):797-802. |
|
Batista MC, Cartledge TP, Zellmer AW et al: Evidence for a critical role of progesterone in the regulation of the midcycle gonadotropin surge and ovulation. J Clin Endocrinol Metab. 1992 Mar;74(3):565-70. |
|
Tasfiri A, Chun SY: Ovulation. In: Adashi EY, Rock JA, Rosenwaks Z (eds): Reproductive Endocrinology, Surgery, and Technology. Philadelphia, Lipincott-Raven, chapter 11: 234, 1996. |
|
Einspanier R, Schonfelder M, Muller K et al: Expression of the vascular endothelial growth factor and its receptors and effects of VEGF during in vitro maturation of bovine cumulus-oocyte complexes (COC). Mol Reprod Dev. 2002 May;62(1):29-36. |
|
Espey LL: Comprehensive analysis of ovarian gene expression during ovulation using differential display. Methods Mol Biol. 2006;317:219-4 1. |
|
Richards JS, Liu Z, Shimada M: Immune-like mechanisms in ovulation. Trends Endocrinol Metab. 2008 Aug;19(6):191-6. Epub 2008 Apr 11. |
|
Duffy DM, Seachord CL, Dozier BL: An ovulatory gonadotropin stimulus increases cytosolic phospholipase A2expression and activity in granulosa cells of primate periovulatory follicles. J Clin Endocrinol Metab. 2005 Oct;90(10):5858-65. Epub 2005 Jun 21. |
|
Tsafriri A, Abisogun AO, Reich R: Steroids and follicular rupture at ovulation. J Steroid Biochem. 1987;27(1-3):359-63. |
|
Ravindranath N, Little-Ihrig L, Phillips HS et al: Vascular endothelial growth factor messenger ribonucleic acid expression in the primate ovary. Endocrinology. 1992 Jul;131(1):254-60. |
|
Sugino N, Suzuki T, Sakata A et al: Angiogenesis in the human corpus luteum: changes in expression of angiopoietins in the corpus luteum throughout the menstrual cycle and in early pregnancy. J Clin Endocrinol Metab. 2005 Nov;90(11):6141-8. Epub 2005 Aug 23. |
|
Wulff C, Wilson H, Rudge JS et al: Luteal angiogenesis: prevention and intervention by treatment with vascular endothelial growth factor trap(A40). J Clin Endocrinol Metab. 2001 Jul;86(7):3377-86. |
|
Fraser HM, Wilson H, Morris KD et al: Vascular endothelial growth factor Trap suppresses ovarian function at all stages of the luteal phase in the macaque. J Clin Endocrinol Metab. 2005 Oct;90(10):5811-8. Epub 2005 Jul 26. |
|
Hutchison JS, Zeleznik AJ: The rhesus monkey corpus luteum is dependent on pituitary gonadotropin secretion throughout the luteal phase of the menstrual cycle. Endocrinology. 1984 Nov;115(5):1780-6. |
|
Hutchison JS, Zeleznik AJ: The corpus luteum of the primate menstrual cycle is capable of recovering from a transient withdrawal of pituitary gonadotropin support. Endocrinology. 1985 Sep;117(3):1043-9. |
|
Duffy DM, Stewart DR, Stouffer RL: Titrating luteinizing hormone replacement to sustain the structure and function of the corpus luteum after GnRH antagonist treatment in rhesus monkeys. J Clin Endocrinol Metab. 1999 Jan;84(1):342-9. |
|
Dickson SE, Fraser HM: Inhibition of early luteal angiogenesis by gonadotropin-releasing hormone antagonist treatment in the primate. J Clin Endocrinol Metab. 2000 Jun;85(6):2339-44. |
|
Van Vugt DA, Lam NY, Ferin M: Reduced frequency of pulsatile luteinizing hormone secretion in the luteal phase of the rhesus monkey. Involvement of endogenous opiates. Endocrinology. 1984 Sep;115(3):1095-101. |
|
Ferin M, Van Vugt D, Wardlaw S: The hypothalamic control of the menstrual cycle and the role of endogenous opioid peptides. Rec Prog Horm Res 40:441, 1984 |
|
Quigley ME, Yen SS: The role of endogenous opiates in LH secretion during the menstrual cycle. J Clin Endocrinol Metab. 1980 Jul;51(1):179-81. |
|
Van Vugt DA, Bakst G, Dyrenfurth I et al: Naloxone stimulation of luteinizing hormone secretion in the female monkey:influence of endocrine and experimental conditions. Endocrinology. 1983 Nov;113(5):1858-64. |
|
Gindoff PR, Jewelewicz R, Hembree W et al: Sustained effects of opioid antagonism during the normal human luteal phase. J Clin Endocrinol Metab. 1988 May;66(5):1000-4. |
|
Wardlaw SL, Ferin M: Interaction between beta-endorphin and alpha-melanocyte-stimulating hormone in the control of prolactin and luteinizing hormone secretion in the primate. Endocrinology. 1990 Apr;126(4):2035-40. |
|
Wehrenberg WB, Wardlaw SL, Frantz AG et al: beta-Endorphin in hypophyseal portal blood: variations throughout the menstrual cycle. Endocrinology. 1982 Sep;111(3):879-81. |
|
Wardlaw SL, Wehrenberg WB, Ferin M et al: Effect of sex steroids on beta-endorphin in hypophyseal portal blood. J Clin Endocrinol Metab. 1982 Nov;55(5):877-81. |
|
Harris TG, Dye S, Robinson JE et al: Progesterone can block transmission of the estradiol-induced signal for luteinizing hormone surge generation during a specific period of time immediately activation of the GnRH surge-generating system. Endocrinology. 1999 Feb;140(2):827-34. |
|
diZerega GS, Hodgen GD: The interovarian progesterone gradient: a spatial and temporal regulator of folliculogenesis in the primate ovarian cycle. J Clin Endocrinol Metab. 1982 Mar;54(3):495-9. |
|
Morales C, Garcia-Pardo L, Reymundo C et al: Different patterns of structural luteolysis in the human corpus luteum of menstruation. Hum Reprod. 2000 Oct;15(10):2119-28. |
|
Stouffer RL: Corpus luteum formation and demise. In: Adashi EY, Rock JA, Rosenwaks Z (eds): Reproductive Endocrinology, Surgery, and Technology. Philadelphia, Lipincott-Raven, chapter 12: 251, 1996. |
|
Vermesh M, Kletzky OA: Longitudinal evaluation of the luteal phase and its transition into the follicular phase. J Clin Endocrinol Metab. 1987 Oct;65(4):653-8. |
|
Groome NP, Illingworth PJ, O'Brien M et al: Measurement of dimeric inhibin B throughout the human menstrual cycle. J Clin Endocrinol Metab. 1996 Apr;81(4):1401-5. |
|
Berga SL, Mortola JF, Girton L et al: Neuroendocrine aberrations in women with functional hypothalamic amenorrhea. J Clin Endocrinol Metab. 1989 Feb;68(2):301-8. |
|
Ferin M: Stress and the reproductive system. In: Physiology of reproduction, Neill JD (ed), Academic Press, chapter 48:2627,2006 |
|
Ferin M: Clinical review 105: Stress and the reproductive cycle. J Clin Endocrinol Metab. 1999 Jun;84(6):1768-74. |
|
Feng YJ, Shalts E, Xia LN et al: An inhibitory effects of interleukin-1a on basal gonadotropin release in the ovariectomized monkey: reversal by a corticotropin-releasing factor antagonist. Endocrinology. 1991 Apr;128(4):2077-82. |
|
Mouri T, Itoi K, Takahashi K et al: Colocalization of corticotropin-releasing factor and vasopressin in the paraventricular nucleus of the human hypothalamus. Neuroendocrinology. 1993 Jan;57(1):34-9. |
|
Battaglia DF, Brown ME, Krasa HB et al: Systemic challenge with endotoxin stimulates corticotropin-releasing hormone and arginine vasopressin secretion into hypophyseal portal blood: coincidence with GnRH suppression. Endocrinology. 1998 Oct;139(10):4175-81. |
|
Shalts E, Feng YJ, Ferin M: Vasopressin mediates the interleukin-1 alpha-induced decrease in luteinizing hormone secretion in the ovariectomized rhesus monkey. Endocrinology. 1992 Jul;131(1):153-8. |
|
Xiao E, Luckhaus J, Niemann W et al: Acute inhibition of gonadotropin secretion by corticotropin-releasing hormone in the primate: are the adrenal glands involved? Endocrinology. 1989 Apr;124(4):1632-7. |
|
Xiao EN, Ferin M: The inhibitory action of corticotropin-releasing hormone on gonadotropin secretion in the ovariectomized rhesus monkey is not mediated by adrenocorticotropic hormone. Biol Reprod. 1988 May;38(4):763-7. |
|
Gindoff PR, Ferin M: Endogenous opioid peptides modulate the effect of corticotropin-releasing factor on gonadotropin release in the primate. Endocrinology. 1987 Sep;121(3):837-42. |
|
Xiao E, Xia-Zhang L, Ferin M: Inhibitory effects of endotoxin on LH secretion in the ovariectomized monkey are prevented by naloxone but not by an interleukin-1 receptor antagonist. Neuroimmunomodulation. 2000;7(1):6-15. |
|
Krasnow SM, Steiner RA: Physiological mechanisms integrating metabolism and reproduction. In: Physiology of reproduction. Neill JD (ed), Elsevier-Academic Pres, chapter 47:2553, 2006 |
|
Warren MP: Eating disorders and the reproductive axis. In: Adashi EY, Rock JA, Rosenwaks Z (eds): Reproductive Endocrinology, Surgery, and Tecnology. Philadelphia, Lipincott-Raven, chapter 50:1039, 1996. |
|
Reichman ME, Judd JT, Taylor PR et al: Effect of dietary fat on length of the follicular phase of the menstrual cycle in a controlled diet setting. J Clin Endocrinol Metab. 1992 May;74(5):1171-5. |
|
Schweiger U, Pirke KM, Laessle RG et al: Gonadotropin secretion in bulimia nervosa. J Clin Endocrinol Metab. 1992 May;74(5):1122-7. |
|
Schreihofer DA, Amico JA, Cameron JL: Reversal of fasting-induced suppression of luteinizing hormone (LH) secretion in male rhesus monkeys by intragastric nutrient infusion: evidence for rapid stimulation of LH by nutritional signals. Endocrinology. 1993 May;132(5):1890-7. |
|
Schreihofer DA, Parfitt DB, Cameron JL: Suppression of luteinizing hormone secretion during short-term fasting in male rhesus monkeys: the role of metabolic versus stress signals. Endocrinology. 1993 May;132(5):1881-9. |
|
Loucks AB, Thuma JR: Luteinizing hormone pulsatility is disrupted at a threshold of energy availability in regularly menstruating women. J Clin Endocrinol Metab. 2003 Jan;88(1):297-311. |
|
Cameron JL: Regulation of reproductive hormone secretion in primates by short-term changes in nutrition. Rev Reprod. 1996 May;1(2):117-26. |
|
Flier JS: Clinical review 94: What's in a name? In search of leptin's physiologic role. J Clin Endocrinol Metab. 1998 May;83(5):1407-13. |
|
Hileman SM, Pierroz DD, Flier JS: Leptin, nutrition, and reproduction: timing is everything. J Clin Endocrinol Metab. 2000 Feb;85(2):804-7. |
|
Cunningham MJ, Clifton DK, Steiner RA: Leptin's actions on the reproductive axis: perspectives and mechanisms. Biol Reprod. 1999 Feb;60(2):216-22. |
|
Nagatani S, Guthikonda P, Thompson RC et al: Evidence for GnRH regulation by leptin: leptin administration prevents reduced pulsatile LH secretion during fasting. Neuroendocrinology. 1998 Jun;67(6):370-6. |
|
Kojima M, Hosoda H, Date Y et al: Ghrelin is a growth-hormone-releasing acylated peptide from stomach. Nature. 1999 Dec 9;402(6762):656-60. |
|
Schneider LF, Warren MP: Functional hypothalamic amenorrhea is associated with elevated ghrelin and disordered eating. Fertil Steril. 2006 Dec;86(6):1744-9. Epub 2006 Oct 30. |
|
Vulliemoz NR, Xiao E, Xia-Zhang L et al: Decrease in luteinizing hormone pulse frequency during a five-hour peripheral ghrelin infusion in the ovariectomized rhesus monkey. J Clin Endocrinol Metab. 2004 Nov;89(11):5718-23. |
|
Tanaka M, Tatebe Y, Nakahara T et al: Eating pattern and the effect of oral glucose on ghrelin and insulin secretion in patients with anorexia nervosa. Clin Endocrinol (Oxf). 2003 Nov;59(5):574-9. |
|
Loucks AB, Mortola JF, Girton L et al: Alterations in the hypothalamic-pituitary-ovarian and the hypothalamic-pituitary-adrenal axes in athletic women. J Clin Endocrinol Metab. 1989 Feb;68(2):402-11. |
|
Pirke KM, Schweiger U, Broocks A et al: Luteinizing hormone and follicle stimulating hormone secretion patterns in female athletes with and without menstrual disturbances. Clin Endocrinol (Oxf). 1990 Sep;33(3):345-53. |
|
Siiteri PK, McDonald PC: Role of extraglandular estrogen in human endocrionolgy. In: Greep RO, Astwood E (eds): Handbook of Physiology; section 7: Endocrinology. Washington DC, American Physiology Society, chapter 28:615,1973 |
|
Kazer RR, Kessel B, Yen SS: Circulating luteinizing hormone pulse frequency in women with polycystic ovary syndrome. J Clin Endocrinol Metab. 1987 Aug;65(2):233-6. |
|
Apter D, Butzow T, Laughlin GA et al: Accelerated 24-hour luteinizing hormone pulsatile activity in adolescent girls with ovarian hyperandrogenism: relevance to the developmental phase of polycystic ovarian syndrome. J Clin Endocrinol Metab. 1994 Jul;79(1):119-25. |
|
Arroyo A, Laughlin GA, Morales AJ et al: Inappropriate gonadotropin secretion in polycystic ovary syndrome: influence of adiposity. J Clin Endocrinol Metab. 1997 Nov;82(11):3728-33. |
|
Berga SL, Guzick DS, Winters SJ: Increased luteinizing hormone and alpha-subunit secretion in women with hyperandrogenic anovulation. J Clin Endocrinol Metab. 1993 Oct;77(4):895-901. |
|
Burger CW, Korsen T, van Kessel H et al: Pulsatile luteinizing hormone patterns in the follicular phase of the menstrual, polycyctic ovarian disease (PCOD) and non-PCOD secondary amenorrhea J Clin Endocrinol Metab. 1985 Dec;61(6):1126-32. |
|
Judd S, Stranks S, Michailov L: Gonadotropin-releasing hormone pacemaker sensitivity to negative feedback inhibition by estradiol in women with hypothalamic amenorrhea. Fertil Steril. 1989 Feb;51(2):257-62. |
|
Karsch FJ, Bittman EL, Foster DL et al: Neuroendocrine basis of seasonal reproduction. Recent Prog Horm Res. 1984;40:185-232. |
|
Xiao E, Xia L, Shanen D et al: Stimulatory effects of interleukin-induced activation of the hypothalamo-pituitary-adrenal axis on gonadotropin in ovariectomized monkeys replaced with estradiol. Endocrinology. 1994 Nov;135(5):2093-8. |
|
Puder JJ, Freda PU, Goland RS et al: Stimulatory effects of stress on gonadotropin secretion in estrogen-treated women. J Clin Endocrinol Metab. 2000 Jun;85(6):2184-8. |
|
Molitch ME: Disorders of the pituitary lactotroph. In: Adashi EY, Rock JA, Rosenwaks Z (eds): Reproductive Endocrinology, Surgery, and Technology. Philadelphia, Lipincott-Raven, chapter 66:1303, 1996. |
|
Soules MR, McLachlan RI, Ek M et al: Luteal phase deficiency: characterization of reproductive hormones over the menstrual cycle. J Clin Endocrinol Metab. 1989 Oct;69(4):804-12. |
|
Stouffer RL: Corpus luteum function and dysfunction. Clin Obstet Gynecol. 1990 Sep;33(3):668-89. |
|
Sheehan KL, Casper RF, Yen SS: Luteal phase defects induced by an agonist of luteinizing hormone-releasing factor: a model for fertility control. Science. 1982 Jan 8;215(4529):170-2. |
|
Xiao E, Xia-Zhang L, Ferin M: Stress and the menstrual cycle: short- and long-term response to a five-day endotoxin challenge during the luteal phase in the rhesus monkey. J Clin Endocrinol Metab. 1999 Feb;84(2):623-6. |
|
Xiao E, Xia-Zhang L, Ferin M: Inadequate luteal function is the initial clinical cyclic defect in a 12-day stress model that includes a psychogenic component in the rhesus monkey. J Clin Endocrinol Metab. 2002 May;87(5):2232-7. |
|
Xiao E, Xia-Zhang L, Vulliemoz N et al: Astressin B, a corticotropin-releasing hormone receptor antagonist, accelerates the return to normal luteal function after an inflammatory-like stress challenge in the rhesus monkey. Endocrinology. 2007 Feb;148(2):841-8. Epub 2006 Nov 2. |
|
De Souza MJ, Miller BE, Loucks AB et al: High frequency of luteal phase deficiency and anovulation in recreational women runners:Blunted elevation in FSH observed during the luteal-follicular transition. J Clin Endocrinol Metab. 1998 Dec;83(12):4220-32. |
|
De Souza MJ: Menstrual disturbances in athletes: a focus on luteal phase defects. Med Sci Sports Exerc. 2003 Sep;35(9):1553-63. |
|
Beitins IZ, McArthur JW, Turnbull BA et al: Exercise induces two types of human luteal dysfunction: confirmation by urinary free progesterone. J Clin Endocrinol Metab. 1991 Jun;72(6):1350-8. |
|
Bonen A: Exercise-induced menstrual cycle changes. A functional, temporary adaptation to metabolic stress. Sports Med. 1994 Jun;17(6):373-92. |